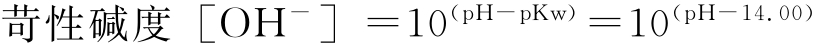能量流动与平衡
第三节 能量流动与平衡
一、水系的组成与性质
1.水的特性与水分子结构
水是最常见的物质,但它有许多异常特性。也正是由于这些特性,才使水在自然界与人类生活中普遍发生巨大作用,成为支配自然与人类环境中各种现象的主要因素。要研究水及其中杂质共同显示的水质特性,需先深入了解水本身的特性。
在地球表面的环境条件下,水可能呈三种物理状态,即液态、气态与固态。由于沸点与冰点间温度范围相当宽,且相变热很大,所以地球表面大量的水还是呈液态,于是组成了各种类型的天然水系。通常条件下呈液态这一点也正是水的最重要特点之一。
水的特性和水的分子结构有关。水分子中的氧原子受到四个电子对包围,其中两电子对和两个氢原子共享,形成两个共价键;另外两对是氧原子本身所持有的孤对电子。四个电子对间由于带负电而互相排斥,使它们有呈四面体结构的倾向,但因孤对电子占据的空间较小,和共享电子对相比具有更大的斥力,因此使H—O—H键角由109.5°(几何正四面体)缩减到104.5°。
氧原子具有比氢原子大得多的电负性,所以水分子中的两共享电子对趋向于氧而偏离氢,于是就在两个孤对电子上集中了更多负电荷,使水分子成为具有很大偶极矩的极性分子。这样的一个水分子就有可能通过正、负电间静电引力和近傍的四个水分子以氢键相联系。分子间氢键力大小为18.81kJ/mol,约为O—H共价键的1/20,冰溶化成水或水挥发成水汽,都首先需要外界供能破坏这些氢键。表明了水分子中氢键的一些结构参数。
当冰开始熔化成水时,冰的疏松的三维氢键结构中约有15%氢键断裂,晶体结构崩溃,体积缩小而密度增大。如果有更多热能输入体系,将引起:①更多氢键破裂,结构进一步分崩离析,密度进一步增大;②体系温度升高,分子动能增加,由于分子振动加剧,而每一分子占据更大体积空间,所以这一因素又使密度趋于减小。上述两因素随温度升高而相互消长的结果,使淡水在3.98℃时有最大密度。这种情况对水生生物越冬生活具有特别重要意义。
在气相中的水极大多数以单分子形态存在,在一般温度与压力条件下,只有少量以二聚体或三聚体的形态存在。
从水的分子结构与水分子有形成较强氢键能力等基本性质出发,还可以解释液态水的其他特性,如“热惰性”、大的表面张力等。
2.天然水系的类别
天然水在自然循环过程中受到污染,混入各种杂质,因此使各种水系具有不同水质。对某一天然水系,可以从地理、地质、物理、化学、生物等方面来描述它的性质状态,但从环境化学角度看问题,应突出这些方面和污染物性质之间的关系。以下我们将就几种重要的天然水系,着重描述它们和环境化学相关的一些性质。
(1)海洋
海洋覆盖着70.8%的地球表面,总面积约3.61×1014m2,平均深度3800m,所以总体积为1.37×1018m3。
海水有很大含盐量,离子强度I=0.7,显示出强电解质溶液性质。对于海水与纯水的各种物理性质。相比之下,海水具有冰点下降特性,且有很大的密度、电导率、折光率、渗透压等。
温度与盐度是决定海水各种性质的决定性因数。海洋中的温度与盐度随深度而变化的情况随溶解氧浓度及浅水区光透过度和深度有一定的关系。海洋表层盐度因海域、降水、蒸发、结冰与融冰等因素而异;表层温度在太阳辐照的日变化与年变化影响下也会发生显著变化。但因水体热容量大,所以温度变化幅度比陆地小得多。就总体来说,海水中盐度可能达到35‰、平均温度不超过4℃、光透过性大约是数十米。海洋表层是富氧的,这起因于大气氧的补充与海中浮游生物的光合作用。在深水地区直到海底氧含量很低又很均一。
海水pH值在表层为8.1~8.3,在深层可下降到7.8。海水中溶解着大量盐类与气体,化学组成十分复杂,几乎包含了周期表中所有的元素。关于海水化学组成问题在此只能作一简述。溶解于海水中的物质按它们存在的数量可分为三类,即主要离子、少量物质与微量元素。占溶质总量99%的主要成分依次为氯离子、钠离子、镁离子、硫酸根离子、钙离子、钾离子与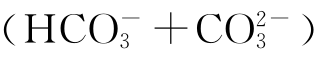 。由于各大洋水流相通,而且混合充分,因此这些主要离子中除(碳酸氢根离子与碳酸根离子)浓度变动较大外,其他离子的相对比例基本上是恒定的(但并不排除海水组成在水平与垂直方向上有规律的一定变化)。因此可通过含氯量来推算其他主要组分在海水中的浓度。
。由于各大洋水流相通,而且混合充分,因此这些主要离子中除(碳酸氢根离子与碳酸根离子)浓度变动较大外,其他离子的相对比例基本上是恒定的(但并不排除海水组成在水平与垂直方向上有规律的一定变化)。因此可通过含氯量来推算其他主要组分在海水中的浓度。
含氯量包括水样中氯化物、溴化物与碘化物总和(后两者被折算为氯化物)。盐度与含氯量之间的经验关系式为:
盐度‰=0.03+1.805(含氯量‰)
盐度在此被定义为在水样中所有碳酸盐转化为氧化物,所有溴化物与碘化物被转换为氯化物以及所有有机物被氧化后计得的水中总固体物浓度。
海水中主要组分的最基本化学参数(离子对离解常数与活度系数)用基本的数据。由这些数据出发,可通过组建化学模型以计算法求得这些组分在海水中相互结合的形态分数。镁离子、钠离子等阳离子与氯离子阴离子在海水中很大程度甚至全部呈自由离子状态存在;碳酸根离子、硫酸根离子、碳酸氢根离子则可和阳离子形成离子对,且各自的离子对形成能力依次递减。
关于海水中的微量金属离子的存在形态问题,由于这些离子大多会发生水解或和氯离子、硫酸根离子、碳酸根离子等配位体形成各种络合离子,且其存在形态还受水域深度、氧化还原电位、生物浓集等因素影响,所以情况要比常量离子复杂得多。虽然在具备必要的数据基础上也能类似常量离子那样作计算处理,但需要再经过分析手段予以验证。可以想见,相关方面的分析技术也有着相当的难度与复杂性。
对海水中溶解性有机碳(DOC)的确切组成尚不了解,其浓度一般为1~5mg/L,近海岸处可能达到20mg/L,而在300米以下深处可降低至0.5mg/L。
显然,海洋是一个开放系统,它时刻和外系统间发生物质与能量的交换。例如,每年有3.3×1013~3.8×1013 m3的水由河流流入海洋,其中带入溶解盐类有38.5亿吨,悬浮物有32.5亿吨。此外在海洋——大气、海洋——海底间也都发生着物质与能量往返传输与交换的过程。在大气与海洋交界的海面能生成微粒气溶胶,湿度较高时,颗粒较大;湿度低时,可呈干的海盐微粒。这些颗粒物可经过大气对流或风力运送达到数千米高空或内陆地区,在大气降水形成过程中起凝结核作用。这些颗粒物的化学组成和海水有很大差异,这是因为:①海面上水泡破裂形成水滴时,因各种气象因素作用,其成分已与海水相异;②海水滴蒸发干涸时,其中容易结晶的组分率先析出分去,结果成为两颗或两颗以上化学组成与性质相异的粒子。
有关海洋污染的问题约有以下六个主要方面:
由汇入海洋的河水夹带的工农业废水以及投弃海洋的各种工业废物;
在由河水夹带入海的生活污水中含有很丰富的营养物质,可在河口、海湾地区引起赤潮;
依傍海岸建立的核电站、热电站排水中的放射性污染物与热污染;
由运输船只机房排出的机油,由海难事件中油轮倾翻的大量原油;
各种塑料制件(破碎鱼网、船上丢弃入海的废塑料制品);
由旅游事业引起的海滨地区污染等。
(2)河流
大气降水及来自地下的水向低洼处汇集、并在重力作用下沿泄水的长条形凹槽流动、且终年有水者称为河流。常年性流水与槽床(即河床)是形成河流的基本条件。关于河流水体的基本综合性质有受纳水量、水位、流速、流量、含固量、矿化度(即以g/kg表示的离子总浓度)等。
和地下水相比,河流是敞开流动水体;和海洋相比,河流只有很小的水量(占地球总水量的百万分之一)。所以河流水质变动幅度很大,因地区、气候等条件而异,且受生物与人类社会活动的影响最大。
一般说来,河水(还有海水)都是含碳酸型的水质系统,以平衡碳酸组分(作为水质的基本调节因素,因此其化学成分也有一定的稳定性。在主要离子中,一般钠离子、钙离子占大多数,阴离子含量一般递减顺序是碳酸氢根离子、氯离子、硫酸根离子。河流的主要污染物是各种有毒金属与各类有机物。
世界上大多数工业城市都是依傍着大的河流建造发展起来的。生产用水与生活用水以及随后产生的废水、污水都以河流作为吞吐对象。也就是说,河流是人们汲取用水的源泉,也是藏垢纳污之处。虽然许多工厂,特别是造纸厂、食品加工厂、化工厂、钢铁厂、石油炼制厂等都设有废水处理系统,但这种系统的处理效率不可能是百分之百的,最终排水中仍还含有一定数量的有毒有害物质。
释入河流的重金属污染物(Hg、Cd、Pb等)很容易被水中悬浮颗粒物吸附,随即沉入水底。所以中上层水体受金属污染的程度较轻微,危害性也较小。当富含有机物的城市污水经过排污管释入河流水体后,随水逐流的污染物会引起上下游水段内溶解氧渐次降低的效应,造成种种不良环境后果。
(3)湖泊
由地面上大小形状不同的洼地积水而成湖泊。形成湖泊必须条件是要具有一个周围高、中间低的能蓄水的湖盆以及长期有水蓄积。湖岸线形态自湖盆形成起就与时俱变,主要因为湖面受风力发生波浪而侵蚀湖岸,由此产生砂土,并沿湖岸流动,再通过湖岸流及下层湖水的返流被搬运到湖深处,从而形成湖棚与湖棚崖。经过长时期地质年代演变,湖盆还有可能被各种来源的砂土或湖内产物所埋没。
湖水水流缓慢,蒸发量大,蒸发掉的水靠河流及地下水补偿。湖水中含钙、镁、钠、钾、硅、氮、磷、锰、铁等元素,其中氮、磷等元素引起的富营养化问题是湖泊的主要污染问题。呈低营养度的水体适宜于水体流动与水生生物游动,而中等营养度的水体最适宜藻类与鱼类等水生生物正常生活,但具有高营养度的水体反而造成藻类大量萌生,水中溶解氧浓度大为降低,因此进一步引起水道阻塞、鱼类生存空间缩小、有害有毒的还原性气体硫化氢的产生等一系列不良后果。还可以认为,富营养化是湖泊等水体的衰老的体现,极端富营养化会使湖泊演化为沼泽或干地。近代,由酸雨引起湖水酸化是湖泊的另一严重污染问题。例如由火成岩基质组成湖盆的湖泊因缺少碱性物质而不能抵御酸雨侵袭,当湖水pH值降到5.5以下时,会发生鱼类大量死亡的后果。
(4)降水
大气降水有多种形态,主要的是雨与雪。当云中水蒸气迅速发生凝结的时候,就发生降水。一般说来,雨水是含杂质少的较洁净的水体。但大气污染及当地地理与气象条件对水质有很大影响。如近海地区雨水中多氯、钠、钙、镁、锶等盐类,而人为污染物含量较少;城市上空的降水可能混入煤烟、工业粉尘等。
降水污染问题一般归入大气环境化学内容。
(5)地下水
地球表面的淡水大部分是贮存在地面之下的地下水,所以地下水是极宝贵的淡水资源。地下水的主要水源是大气降水。降水中一部分通过土壤与岩石的间隙而渗入地下形成地下水。严格地说,存在于地表之下饱和层的水体才是地下水。
降水抵达地面之后,在和土壤、岩石物质及细菌等长久反复接触的天然过程中,发生了过滤、吸附、离子交换、淋溶与生物化学等作用,使原降水水质发生很大变化。归纳起来,地下水水质有如下特点:①悬浮颗粒物含量很少,水体清沏透明;②无菌、盐分高、硬度大、含较多量的有机物;③不和空气接触,水体呈还原态,铁、锰等元素以亚铁离子、锰离子低价形态存在;④水温不受气温影响;⑤因有岩石等阻隔,流动速度很小,各部位水层的水质也可有很大差异。
地下水中污染物质主要来源于人们的生产或生活活动,具体说来有这样一些方面:
耗氧污染物。当某些生活污水、工业废水或固体废物的沥取液流过土壤表层时,其中一些耗氧物质被土壤过滤或者发生生物氧化而被除去,但其残留部分仍可能进入地下水水体;
病原体,如细菌、病毒、原生物动等。污水流过土壤层后,细菌与较大个体的微生物被滤除;病毒虽能穿透土层,但进入地下水水体后,经长时间迁移会失去活性。但在一些选址不当的浅井井水中仍然有可能检出这些病菌;
植物营养物质。当降水流经过富含肥料的土壤时,土壤中所含的氮、磷化合物可能被淋溶而穿透土层,随后归入地下水水体;
有机化学物品。例如在低浓度下就呈现较大毒性的杀虫剂、农药随农田灌溉水渗滤而进入地下水体;
放射性物质。随着原子能工业的发展,世界范围内积累了大量的放射性废弃物,其处置法之一就是深井投弃。经过投弃的放射性废物经年长日久之后,难保不会渗入地下水水体,尤其是长寿命的放射性核素对地下水的危害性更大。
总的说来,地下水不如地面水那样容易受到污染,但因为地下水基本上属于封闭水系(深层地下水的滞留时间可达几千年),在地层之下不易挥发、不被稀释与不易发生降解,因此水系一旦受到污染,就十分难以通过自然过程或人为手段予以消除。除污染问题外,过量汲取与利用地下水将会引起海水倒灌入井与陆地沉降等问题。
3.天然水体中化学物质的存在形态
化学物质在环境中有一定的赋存形态。广而言之,“形态”一词含义包括物理结合状态、化学态(有机的或无机的)、价态、化合态与化学异构态等多方面。
具有一定形态的化学污染物在环境中有其发生与演变的过程。认为污染物具有确定的分子结构与环境特性,只是相对的,而其变化则是绝对的。例如进入环境的甲基汞在不同环境介质间迁移或与各种环境因子相互作用的过程中,甲基汞的“母体形态”(CH3Hg+)具有相对稳定性。
在不同的环境介质中,甲基汞随其所依附基体的不同而呈现各异的“基体形态”。如在水中甲基汞的基体形态为[CH3Hg(OH)],当其迁移转入大气、土壤或生物组织之后,它的形态就相应地转化为[CH3HgCH3]、[CH3Hg-腐植质]或[CH3Hg-S-质]。
此外,在作甲基汞浓度分析时,还需要将它在样品中的基体形态转化为某一稳定的、可为仪器响应的“分析形态”CH3HgCl,而后送入仪器进行测定。
认定环境污染物在一定时限内的存在形态,并掌握它在环境因素影响下所发生形态变化,相关方面的研究有很大环境化学意义。
对此,列举三个例子略加说明。
一是对于水体中浮游生物与鱼类来说,游离的金属离子形态可能是最富毒性的,稳定络合物或与胶粒相结合的形态则是低毒或无毒的。例如由酸雨引起湖水酸化,使原先存在于水体中的聚合氢氧化铝胶体转化为可和鱼鳃粘膜反应的水合铝离子形态,从而破坏了生物膜的正常功能,可导致鱼类大量死亡;
二是存在于海水中铜的可能形态有铜离子、碳酸铜、铜的氢氧化物、铜的氯化物、次氯酸铜、硫酸铜等,总浓度约2×10-3mg/L,用原子吸收分光光度法测定铜的总浓度与用离子选择性电极法测定离子态铜离子的浓度,两方面的结果显然是不可比拟的。通过这个例子可使我们认识到,在对水体中某些元素制订其监测方案与选定分析方法时,必须事先掌握相关研究对象存在形态方面的知识;
三是用离子交换或螯合萃取等方法对含金属工业废水处理的效率,完全取决于废水中金属呈离子形态的含量分数。
4.天然水体中的异相物质
(1)水体中的各种沾染物
化学意义上的纯水只能在实验室中才能制得,而任何一种天然水体都不是纯水体系。即使是非污染水体,也含有许多种类与数量不一的杂质(沾染物)。
天然水体中的杂质按其密度差异,可分别分散在上、中、下三个层次之中。在和空气交界的界面层富含脂肪酸、酯类等化合物,这些物质的分子大多是疏水性的,是造成这一层水表面张力增大的原因。
因此,一些微小水生动物(如水蜘蛛)能在这类似绷床的表层水面上活动与生活。这一层之下还有一个含多糖、蛋白质等营养物质的薄层。在这里,溶解性有机物质浓度可达2~9g/L,所以是水中微生物的一个重要活动场所,每毫升水中可含108个细菌个体。在上层水面,还可能飘浮着各种生活垃圾(木片、纸屑等)与多种藻类。作为水体的另一个重要组成部分,在底层的沉积物中含有各种颗粒度不等的砾、砂、粘土、淤泥、生物的排泄物与尸体以及各种天然与人造的化学物质(金属、颗粒状有机物等)。
在占极大体积比率的中间层中所含的杂质主要是溶解性的分子与离子、胶体微粒与悬浮颗粒物。这些杂质的来源与颗粒度大小分别。溶解性分子与离子的粒度一般不大于10-3μm,这类组分不可能通过过滤或沉降的方法从水中分去。胶体粒子可通过丁达尔效应观察其在水体中的存在,这类粒子能穿过大多数过滤介质的孔目,且沉降速度甚慢。直径大于1微米的悬浮颗粒能被一般过滤介质滞留,也能在水中迅速沉降。这些颗粒能阻碍日光透过,是造成水体外观混浊的原因。水体中大多数微生物也属此类。
天然水体中的生物种类与数量多得不可胜数,但可简单地划分为底栖生物、浮游生物、水生植物与鱼类四大类。生活在水体中的微生物是关系到水质的最重要的生物体,对此又可分为植物性的与动物性的两类。植物性微生物按其体内是否含叶绿素又可分为藻类与菌类微生物。
一般的细菌(单细胞与多细胞)与真菌(霉菌、酵母菌等)都属于体内不含叶绿素的菌类。生活在水体中的单细胞原生动物以及轮虫、线虫之类的微小动物都是动物性的微生物。生活在天然水体中的较高级生物(如鱼)在数量上只占相对很小的比例,所以它们对水体化学性质的影响较小。相反,水质对它们生活的影响却很大。以下对水体中悬浮颗粒、细菌与藻类这几种异相物质的相关方面作进一步阐述。
(2)水体中的颗粒物质
取一份水样,根据一定的程序操作,可测得水样中相关悬浮固体物与溶解固体物的各种参数。总残渣包括过滤性残渣(又称总溶解性固体物TDS)与非过滤性残渣(又称悬浮物或悬浮固体物SS),后者是表征多种地表水与废水水质的重要物性参数,成为水质监测中的必测项目。总残渣又可分为挥发性残渣与固定残渣两部分,它们分别和水样中有机物与无机矿物质含量有关。
天然水体中粒子物质的来源与相应的性质主要有如下五个方面:
陆生的岩石碎屑,包括性质稳定的石英(SiO2)、刚玉(Al2O3)或赤铁矿(Fe2O3)等颗粒物。它们是由陆地岩石经过各种风化作用碎裂以后,再经过水流、风或海滩波浪作用进入水体。它们密度较大、容易沉降;颗粒的粒级范围较宽,但在某一窄小的沉积区域中有较整齐的粒度。
粘土矿物,原是土壤的主要成分铝硅酸盐经过风化作用生成,再经过风或水流载带进入各水体。其中所含水合金属氧化物是水解产物,可能通过凝聚作用而沉降。
碳酸盐与硅石,是由生物体残骸中“硬性”部分经过分散而形成,在营养物供给充分的水体中含量较丰富。通过各种复杂的自然过程,这些颗粒物又常以珊瑚礁等形态聚集在水体浅底部位。
颗粒状有机物,是生物体残骸中“软性”部分,其中包含生物体碎屑与相当多数量细菌,由它们构成了水体中大多数的悬浮颗粒物。
这一类物质中只有很小部分能组成水底沉积物,但在别的沉积物迁移与变化过程中起很大作用。
天然与人造的化学物质,这类物质种类众多,含量变化也较大。天然化合物多数是一些大分子的碳水化合物、类脂物与蛋白质;人造的包括有机农药、糖脂、重金属水解产物、放射性核素及颗粒状有机物(如纸浆厂排水中的含有物)等。
天然水体中各种颗粒物有不同的浓度与不同的滞留时间,它们在生成过程与迁移过程中受到重力、水力(水平与垂直方向)以及相互之间的吸引力,最后以沉积物形态汇集于水底。
(3)藻类
藻类是在缓慢流动水体中最常见的浮游类植物。按生态观点看,藻类是水体中的生产者,它们能在阳光辐照条件下,以水、二氧化碳与溶解性氮、磷等营养物为原料,不断生产出有机物,并放出氧。合成有机物一部分供其呼吸消耗之用,另一部分供合成藻类自身细胞物质之需。在无光条件下,藻类消耗自身体内有机物以营生,同时也消耗着水中的溶解氧,因此在暗处有大量藻类繁殖的水体是缺氧的。
按藻类结构,它们可能是以单细胞、多细胞或菌落形态生存。一般河流中可见到的有绿藻、硅藻、甲藻、金藻、蓝藻、裸藻、黄藻等大类,它们的外观大多数有鲜明的色泽,这是因为在它们的体内除含叶绿素外,还含有各种附加色素,如藻青蛋白(青色)、藻红蛋白(红色)、胡萝卜素(橙色)、叶黄素(黄色)等。水体中藻类种类与数量可依季节与水体环境条件(底质状况、含固量、水速、水污染状况等)而有很大变化。
藻类等浮游植物体内所含碳、氮、磷等主要营养元素间一般存在着一个比较确定的比例。按质量计算,大约是C∶N∶P=41∶7.2∶1,按原子数计算,大约是C∶N∶P=106∶16∶1。大致的化学结构式为(CH2O)106(NH3)16H3PO4。藻类大量繁殖是水体富营养化的标志,由此可从多方面影响水体的水质。
(4)细菌
细菌是关系到天然水体环境化学性质的最重要生物体。它们结构简单、形体微小,在环境条件下繁殖快分布广。
就生态观点看,它们中多数是还原者。由于比表面甚大,从水体摄取化学物质的能力极强,还由于细胞内含有各种酶催化剂,由此引起生物化学反应速度也十分快。
按外型可将细菌分为球菌、杆菌与螺旋菌等类。它们可能是单细胞或多至几百万个细胞的群合体。细胞体表面荚膜层由多糖或多肽类化合物构成,具有保护自身免受其他微生物进攻的作用。
在荚膜层上还联结着很多基团(羧基、氨基、羟基等),所以在水体pH发生变化时,可能通过这些基团的电离或质子化作用等使细胞体表面带电:
低pH条件下+H3N(+Cell)CO2H(带正电)
中pH条件下+H3N(Cell)COO(不带电)
高pH条件下H2N(-Cell)COO(带负电)
按营养方式,可将细菌分为自养菌与异养菌两类。自养菌具有将无机碳化合物转化为有机物的能力,光合细菌(绿硫细菌、紫硫细菌等)与化能合成细菌(硝化菌、铁细菌、氢细菌、硫氧化细菌等)属于此类。大多数细菌属于化能异养型,它们合成有机物的能力弱,需要现成有机物作为自身机体的营养物。
异养菌又分为腐生菌与寄生菌。前者包括腐烂菌、放线菌等,它们从死亡的生物机体中摄取营养物;寄生菌则生活在活的机体中,一些病原性细菌属于此类,它们以进入水体的生物排泄物为媒介,传播各类疾病。
按照有机营养物质在氧化过程(即呼吸作用)中所利用的受氢体种类,还可将细菌分为:①好氧细菌,如醋酸菌、亚硝酸菌等。这类菌体生活在有氧环境中,以氧分子(大气中氧或水体中溶解氧)作为呼吸过程中受氢体;②厌氧细菌,如油酸菌、甲烷菌等。这类菌体只能在无氧环境中(土壤深处、生物体内)呼吸、生长与繁殖,呼吸过程中以有机物分子本身或二氧化碳等作为受氢体;③兼氧细菌,如乳酸菌等。这类细菌兼能在有氧或无氧条件下进行两种不同的呼吸过程。
菌体的主要组成物质是水(约占80%),其余部分为有机物质与少量无机物质(约分别占18%与2%),前者的化学组成可用近似经验式C5H7O2N表示,所含无机物质包括磷、铁、硫等化合物。
如前已述及,表面飘浮层是水体细菌麇集的地方,实际上在水体中的各种相界面大多是营养物质富集之处,这些界面都可作为微生物很好生长繁殖的生境。
二、水系中的化学平衡
1.气态物质与液态物质在水中的溶解平衡
(1)气体在水中的溶解
在水体中的溶解性气体对水生生物类有很大的意义。例如鱼类在水体中生活时,要从周围水中摄取溶解氧(溶解氧小于4mg/L时就不能生存),经过体内呼吸作用后,又向水中放出二氧化碳。对于水中藻类来说,则是通过其体内进行的光合作用,有着和呼吸作用相反的过程。又如水体中溶解氮量因某些原因增大时,会引起水中大量鱼类与其他水生生物死亡。当然,许多工业排气,如氯化氢、二氧化硫、氨气等一旦进入水体并进一步溶解之后,也会对水体产生各种不良的影响。
溶解平衡是相对的,而偏离平衡状态的水中溶解气体(处于不饱和或过饱和状态)有在气-水两相间发生传质的趋向,由此关系到气体物质在两环境圈层间发生迁移的过程。
能溶于水并形成电解质或非电解质溶液的气体,它们的溶解度都可以用亨利定律来表述。亨利定律的内容是:“在定温与平衡状态下,一种气体在液体里的溶解度与该气体的平衡压力成正比”。用公式表示为:
[A(aq)]=KHApA
式中A——代表某种气体;
pA——分压;
KHA——亨利系数,在一定温度下KHA是常数。
在应用亨利定律时须注意下列几点:
溶质在气相与在溶剂中的分子状态必须相同,否则便不能使用亨利定律。例如二氧化碳溶解在水中时,经过水合、电离作用后,在水中有多种存在形态:水合二氧化碳、碳酸、碳酸氢根离子、碳酸根离子,亨利定律表达式中[A(aq)]只包含水合二氧化碳这一形态。
对于混合气体,在压力不大时,亨利定律对每一种气体都能分别适用,和另一种气体的压力无关。
对于亨利常数大于10-2的气体,可认为它基本上是能完全被水吸收的。
亨利常数作为温度的函数,有如下关系式:
dlnKH/dT=△H/RT2
式中,△H为气体溶于水过程的焓变。一般△H为负值,所以随温度降低,亨利系数增大,即低温下气体在水中有较大溶解度。对于溶解度十分大的气体,亨利系数还可能和浓度相关。
亨利常数的数值可以在定温下由实验测定,也可以使用热力学方法予以推算。(https://www.daowen.com)
亨利定律有几种不同表达式,应用时要注意辨别。
(2)氧在水中的溶解
氧在水中的溶解度与溶解氧值是两个既相区别而又相联系的概念。氧在水中的溶解度指的是水体与大气处于平衡时氧的最大溶解浓度,它的数值和温度、压力、水中溶质量等因素相关。水中溶解氧值则一般是指非平衡状态下的水中溶解氧的浓度。它的数值和水体曝气作用、光合作用、呼吸作用及水中有机污染物的氧化作用等因素相关。这两个概念之间的差异是由于大气与水体界面间氧气传质动力过程较慢而引起的。
①氧在水中的溶解度
若已知当25℃下水蒸气在空气中含量为0.0313摩尔分数以及干空气中含20.95%O2时,则可应用道尔顿分压定律与亨利定律算出标准条件下氧在水中溶解度[O2(aq)]:
[O2(aq)]=Ko2·po2=1.28×10-8×(1.0000-0.0313)×1.013×105×0.2095
=2.63×10-4mol/L(相当于8.4mg/L)
由上式可导出在定压条件下温度对氧气在水中溶解度影响的关系式:

式中C1与C2——分别为绝对温度T1与T2下气体在水中溶解度(mg/L);
△H——溶解热(J/mol);
R——气体常数(8.314J/K·mol)。
②水中溶解氧(DO)值
水中溶解氧值是水质的重要参数之一,也是鱼类等水生动物生存的必要条件。由于各种环境因素的影响,水中DO值变化很大,即在一天之中也不相同。主要的影响因素有:再曝气作用、光合作用、呼吸作用与有机污染物的氧化作用。再曝气作用和水中DO值相关,当DO值和水中氧的溶解度差值越大时,氧从空气进入水中的量也越多。澎湃奔流的河水由于和空气交界面积较大,再曝气作用的过程进行得较快。
水中植物体的光合作用在白昼进行,由于过程中产生氧气,也使水中DO值增大。水中各类生物体的呼吸作用是全天不分昼夜地进行,并不断从水中耗用氧而使DO降低。早晨日出后,由于光合作用与再曝气作用同时发生,水中DO值不断上升;但过了午后,因DO值超过了溶解度,以致再曝气过程发生逆转,氧反而从水中释出,因而曲线开始下降。傍晚日落后光合作用停止,因此曲线继续下降。
当水体污染程度较低时,好氧性细菌使有机污染物发生氧化分解而逐渐消失,因此DO值降低到一定程度后不再下降。但如污染比较严重,超出水体自净化能力时,则水中溶解氧耗尽,从而发生厌氧性细菌的分解作用,同时水面常会出现粘稠的絮状物使之与空气隔开,妨碍再曝气作用的进行。
(3)二氧化硫在水中的溶解
二氧化硫与二氧化碳在水中溶解情况有很多相似之处,对于后者将在下一节酸碱平衡中再作详述,在此仅讨论二氧化硫在水中的溶解(所讨论的内容与所得结果大体适用于二氧化碳)。
二氧化硫是一种重要的大气污染物,它的气-液溶解平衡在形成酸雨的问题上有很大意义。
根据电中性原理:
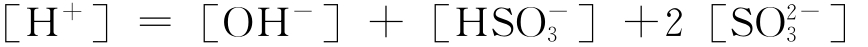
将此式和上列[HSO-3]、[SO2-3]等表达式相联,可得:
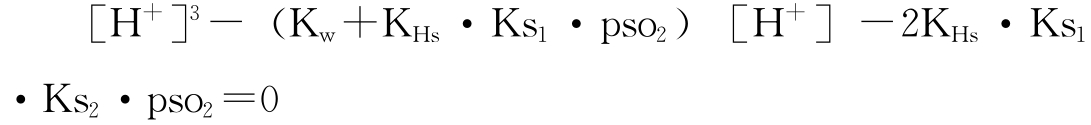
式中Kw为水的离子积。纯净去离子水和假想只含二氧化硫的空气达到平衡时,该含酸水溶液的pH值可通过解以上的三次方程求得。将解得的[H+]浓度值代入上列相关方程,还可求得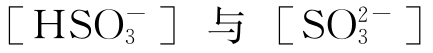 的平衡浓度。
的平衡浓度。
(4)一氧化氮与二氧化氮在水中的溶解
一氧化氮(NO)与二氧化氮(NO2)在水中溶解的系列反应为:

硝酸是强酸,在水溶液中基本上只以硝酸根离子形态存在;亚硝酸是弱酸,它的电离程度由pH值所左右,在水溶液中通常有亚硝酸根离子与HNO2(aq)两种存在形态。以下,我们来考虑在气-液平衡条件下,水相中硝酸、亚硝酸以及它们离子形态浓度和pNO2、pNO之间的函数关系。
2.液态物质在水中的溶解
关于液态物质在水中溶解平衡的规则大多是定性的与经验性的。一般地说,低极性分子组成的物质或分子中不带有能形成氢键的基团的物质在水中溶解度很小。例如,和醇类具有高度水溶性的性质相反,烃类或卤代烃在水中溶解度是很小的。结构相近而分子较小的物质一般有较大的水溶性,例如苯、甲苯、邻二甲苯三者在水中溶解度递降,分别为1.8、0.51、0.17g/L。至于苯酚、苯、环己烷,虽然它们的分子大小相近,但由于极性有较大差异,所以在水中溶解度分别为70、1.8与0.05g/L。
很多呈液态的有机物质在水中依靠分子间作用力发生物理性溶解。
这种分子间作用力有两类:一类是范德华引力,这种力较小,存在于任何分子间;另一类是氢键力,可表示为AH…B,这种力较大,对溶解度有决定性意义。在氢键的表达式中,A与B为电负性大,半径小的原子,如氧、硫、氟、氯、氮等;从电子授受能力来说,AH为受电子基,如—OH、=NH、—SH等;B为给电子基,比如说—O—、=N—、—F、—S—、—Cl等。基于上述,我们可以将溶剂分为以下五种类型:
N型是惰性溶剂,它们没有生成氢键的能力。如苯、甲苯、煤油、硅油、石蜡油、四氯化碳、二硫化碳、环己烷、戊烷、己烷、庚烷等。
A型是受电子型溶剂,分子中含AH基,能与给电子溶剂B的分子生成氢键。如氯代甲烷、氯代乙烷等。
B型是给电子型溶剂,分子中含B原子,能与另一种溶剂AH的分子生成氢键。如各类醚、酮、醛、腈、酯、季胺、吡啶等。
AB型是给受电子型溶剂,分子中同时含有AH基与B原子,因此,自身就可缔合成多聚分子。诸如水、多酚、多元醇、多元羧酸类化合物,它们自身缔合成多聚分子是通过交链氢键键合完成的,可定名为AB1型溶剂。
另有一类AB2型溶剂,诸如醇、酚、伯胺、羧酸类化合物,它们自身缔合成多聚分子是通过直链氢键键合完成的。
三、酸碱平衡
酸碱反应不存在动力学阻碍,多数反应能在瞬间完成,所以仅涉及平衡问题。pH值是体系中最为重要的特性参数,由pH值大体决定体系内各组分的相对浓度。
在天然水环境中,重要的一元酸碱体系有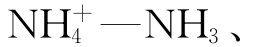
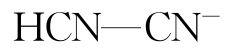 等,重要的二元酸碱体系有
等,重要的二元酸碱体系有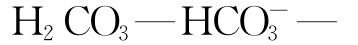
 等,重要的三元酸碱体系有
等,重要的三元酸碱体系有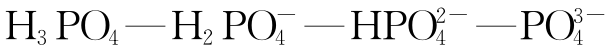 等。强酸或强碱在天然水体中出现的可能很小。
等。强酸或强碱在天然水体中出现的可能很小。
1.天然水的酸碱性
大多数含有矿物质的天然水,其pH值一般都在6~9这个狭窄的范围内,并且对于任一水体,其pH值几乎维持恒定。在和沉积物的生成、转化及溶解等过程相关的化学反应中,天然水的pH值具有很大意义,它往往能决定转化过程的方向。
生物活动,如光合作用与呼吸作用,以及物理现象,如自然的或外界引起的扰动伴随着曝气作用,都会或增或减地使水中溶解性二氧化碳浓度发生变化,从而影响水体pH值。
此外,其他一些借助于生物进行的反应也会影响天然水的pH值,如黄铁矿被氧化的反应会导致pH降低;反硝化或反硫化等过程则趋向于使pH值升高。
在大多数天然水中都有碳酸氢根离子与碳酸根离子作为碱存在。有时还可存在别的低浓度碱,如铍酸根离子、磷酸根离子、砷酸根离子、氨气、硅酸根离子等。火山与温泉向水中加入氯化氢与二氧化硫之类气体,可强烈地产生酸性水。工业废水中含有的游离酸或多价金属离子经过排放而进入天然水系,也可使水具酸性。另外一些酸性成分是硼酸、硅酸与铵离子。总的说来,天然水体中最重要的酸性成分还是二氧化碳,它和水形成相对平衡的碳酸体系。
2.酸——碱体系的特性
(1)酸碱质子理论
在各种酸碱理论中,由布朗斯特德与劳莱于1923年提出的酸碱质子论是最适用于水体化学的一种理论。按这种理论对酸与碱所下的定义是:酸是一种质子给体,碱是一种质子受体。
例如,在下列反应中:

当反应自左向右进行时,盐酸起酸的作用(质子给体),水起碱的作用(质子受体)。由于质子非常微小又不能独立存在,所以在水体中往往由溶剂水分子作质子受体而生成水合氢离子H3O+。
虽然上述反应朝右侧进行的趋势非常强烈,但仍可将反应视为可逆的。如果反应逆向进行,则应将H3O+视为酸,Cl-则为碱。HCl-Cl-与H3O+-H2O实质上是两对共轭酸碱体。
另举一酸碱反应例:

对这个反应来说,水起了酸的作用。比较上述两个例子,可以说,水具有两重性,即它在反应中既能作酸又能作碱,全视它和什么物质作用而定。也可以说,一种物质到底是酸还是碱,脱离了具体的反应是无法确定的。
从酸碱质子理论看来,任何酸碱反应,如电离、中和、水解等都是两个共轭酸碱对之间的质子传递反应。水体中的水分子在这些过程中经常充当质子转移的中间介质。
表按质子论定义的常见酸与碱,不但一般分子可以成为酸或碱,各种正离子与负离子也可以成为酸或碱。
有机酸碱大多是分子态化合物。作为质子给体(酸)的有酸、酚、醇、腈、酰胺等类化合物,作为质子受体(碱)的有醚、酯、酮、叔胺等类化合物。
(2)酸与碱的强度
酸与碱的强度分别用酸电离常数Ka与碱电离常数Kb表示。
相应地有以上Ka、Kb表达式中已将[H2O]浓度项分别并入Ka与Kb,这是因为水在体系中过量存在,它的浓度没有发生显著变化。还应指出,准确的Ka或Kb应由活度来计算,但在十分稀的溶液中基本上可用浓度来代替。
由①与②式可见,酸与碱的强度都是相对于水的共轭体系(H3O+-H2O)与(H2O-OH-)来衡量的。
为了使用方便,一般将Ka、Kb分别转写为pKa、pKb:
pKa=-lgKa ①
pKb=-lgKb ②
Ka数值越大或pKa数值越小,则形态为HA的酸越强。HIO3的pKa=0.8,一般定pKa<0.8者为强酸。Kb数值越大或pKb数值越小,则形态为A-的碱越强。H2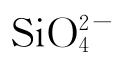 的pKb=1.4,一般定pKb<1.4者为强碱。
的pKb=1.4,一般定pKb<1.4者为强碱。
对于共轭酸碱HA与A-来说,将①、②两反应方程相联,得
Ka·Kb=[H3O+][OH-]
两者之积称为水的离子积Kw,在25℃时
Kw=Ka·Kb=10-14.00
或pKa+pKb=14.00
上式显示,共轭体系中的酸越强,则其共轭碱越弱,否则反然。以下列举一些共轭酸碱对及它们的强度次序。
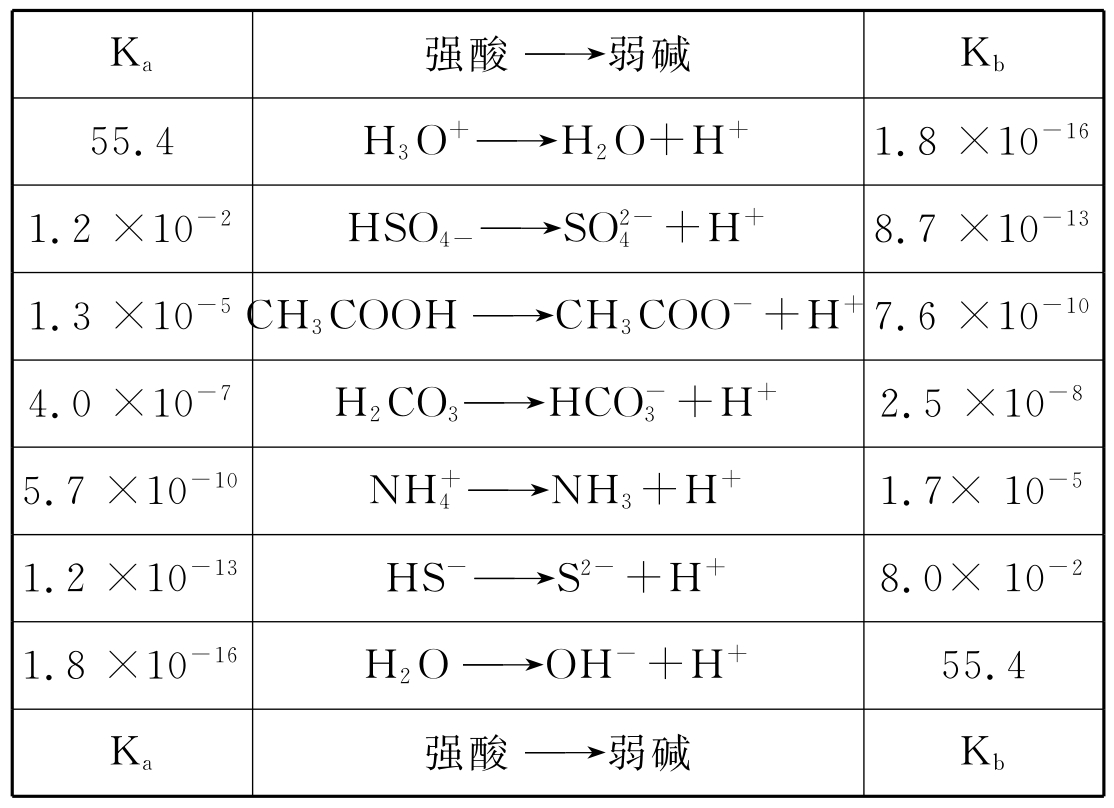
共轭酸碱的相对强度确定后,那么反应按什么规律进行呢?对于下列反应,A1与B1以及A2与B2为共轭酸碱,若它们的强度是A1>A2,B2>B1,则反应从左向右进行。
3.酸碱缓冲容量
向共轭酸碱体系外加别的酸[H+]或碱[OH-],并经酸碱反应被体系消耗后,仅能引起pH值微小变化的事实引出缓冲作用的概念。酸碱体系的缓冲容量(β)被定义为:若使该水溶液体系的pH值升高一个单位所需加入强碱氢氧化钠的摩尔数(cB,mol/L)。β总是取正值,所以若向溶液中加入强酸(cA,mol/L),则相当于取出了同样数量的强碱。
对地面水来说,一般只有很小缓冲容量,不能随意多量受纳酸碱废水,否则将引起pH值很大变化,对水质与水生生物会产生很大影响。
4.碳酸平衡
在大气中含有一定分压的二氧化碳,因此在所有的天然水体中都有相当高浓度的[CO2(aq)]、[H2CO3]、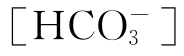 与
与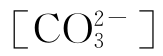 。此外,考虑到二氧化碳在光合作用与呼吸作用中具有重大的意义,则可以想见,碳酸系统的平衡性质在调节天然水体的pH中起着十分重要的作用。除了源于空气中的二氧化碳外,水中碳酸化合物的来源还有岩石、土壤中碳酸盐与重碳酸盐矿物的溶解、水生动植物的新陈代谢、水中有机物的生物氧化等。此外,水质处理过程中有时也需加入或产生出各种碳酸化合物。
。此外,考虑到二氧化碳在光合作用与呼吸作用中具有重大的意义,则可以想见,碳酸系统的平衡性质在调节天然水体的pH中起着十分重要的作用。除了源于空气中的二氧化碳外,水中碳酸化合物的来源还有岩石、土壤中碳酸盐与重碳酸盐矿物的溶解、水生动植物的新陈代谢、水中有机物的生物氧化等。此外,水质处理过程中有时也需加入或产生出各种碳酸化合物。
对于二氧化碳-水系统作和二氧化硫-水系统类同的处理。在不同温度下,二氧化碳的亨利常数KHC与碳酸的一级、二级电离平衡常数Kc1与Kc2的数值也不同,下面对此作若干必要的说明。
①在水溶液中分子状态二氧化碳与碳酸之间存在着如下平衡:
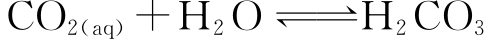
达到平衡时以CO2(aq)存在形态为主,而碳酸形态只占游离碳酸总量中很小比例。如在25℃温度下,[H2 CO3]/[CO2(aq)]=10-2.8。因此将水中游离碳酸总量用[H2CO3]。
在亨利定律表达式中也就可用[H2CO3]来替代[CO2(aq)],这样处理能为平衡计算带来方便。
②一级电离常数Kc1表达式中的分母项用[H2CO3]代替[H2CO3],Kc1被称为实用一级电离常数。
③体系中能接受质子的各种组分相加合构成水样的碱度;能给出质子的各种组分相加合构成水样的酸度。按此定义,酸度与pH值间、碱度与pH值之间,在概念上显然是存在区别的。
以下拟将体系分为封闭的碳酸体系与开放的碳酸体系来作进一步的介绍。
(1)封闭碳酸体系
假定将水中溶解[H2CO3]看作为不挥发酸,由此构成的是封闭碳酸体系。我们可在海底深处、地下水、锅炉水及实验室水样中遇到这样的体系。
在平衡体系中,[H2CO3]、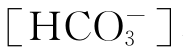 、
、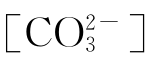 三种形态的浓度比率。
三种形态的浓度比率。
由此三式并通过计算可了解αH2CO3、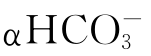 、
、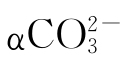 随pH值变化而消长的情况。
随pH值变化而消长的情况。
在低pH区内,溶液中只有二氧化碳与碳酸,在高pH区则只有碳酸根离子,而碳酸氢根离子在中等pH区内占绝对优势。三种碳酸形态在平衡时的浓度比例和溶液pH值有完全相应的关系。每种碳酸形态浓度受外界影响而变化时,将会引起其他各种碳酸形态的浓度以及溶液pH值的变化,而溶液pH值的变化也会同时引起各碳酸形态浓度比例的变化。由此可见,水中的碳酸平衡同pH值是紧密联系的。
综上所述,对封闭碳酸平衡体系的特性归纳如下:
①系统pH值范围约为4.5~10.8。当水样中另外含有强酸时,pH值将小于4.5;或当另外含有强碱时,pH值当可大于10.8。
②pH=8.3的特征点可看作是一个分界点。当体系pH小于8.3时,可以认为碳酸根离子含量甚微,水中只有二氧化碳、碳酸与碳酸氢根离子,可只考虑一级碳酸平衡即:
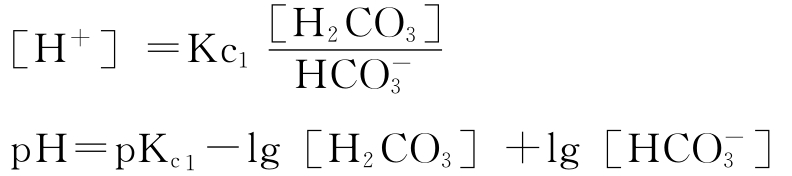
当溶液的pH>8.3时,[H2CO3]的浓度就可忽略不计,认为水中只存在碳酸氢根离子与碳酸根离子,应考虑二级碳酸平衡即:
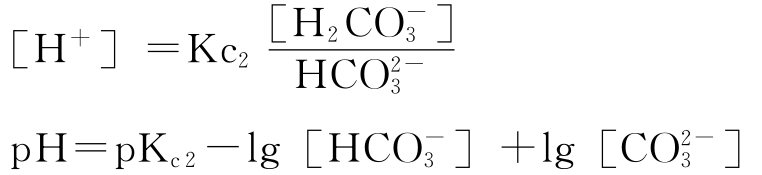
(2)开放碳酸体系
这里所指的是和大气相通的碳酸水溶液体系。按亨利定律,水溶液中碳酸浓度随大气中二氧化碳分压而变:
5.酸度与碱度
天然水体中存在着多量弱酸(如碳酸、硅酸、硼酸、乙酸等),强酸弱碱盐(如硫酸铝、氯化铁等),有时还可能出现某些强酸(如盐酸、硫酸、硝酸等),它们都对水系统提供酸度,其酸度值决定于这些组分的数量与它们的离解程度。可以将总酸度分为离子酸度与后备酸度两部分,前者由H+质子提供并和水样的pH值相应,后者和水系统的缓冲能力有关。
天然水体中还存在着多量弱碱(如氨气、苯胺等),强碱弱酸盐(如碳酸盐、碳酸氢盐及硼酸、硅酸、磷酸、乙酸、腐植酸等的盐类),有时还可能出现某些强碱(如氢氧化钠、氢氧化钙等),它们都对水系统提供碱度,其碱度值决定于这些组分的数量与它们的离解程度。
碱度被定义为:凡在水中离解或水解后生成可与强酸(H+)反应的物质总量称为水的碱度。相应地定义酸度为水中能和强碱(OH-)反应的物质总量。考虑到水中很多物质(如碳酸氢根离子是其一)兼能和强酸与强碱发生反应,碱度与酸度在定义上有交互重叠部分,所以除了pH<4.5的水样外,一般使用了碱度就不再用酸度表示水样的酸碱性。
碱度与酸度是水体缓冲能力的测度,天然水体可受纳酸碱废水的容量受这类参数的制约。此外,各类工业用水、农田灌溉水或饮用水等都有一个适宜的碱度(或酸度)数值范围。
以下将天然水体近似看作纯碳酸体系,对它的酸度与碱度作进一步论述。向含碳酸的清水中加入强酸或强碱,一方面可引起溶液pH值改变,另一方面也促使碳酸平衡向左或向右移动,这样就引起碳酸存在形态的转化。一般清水中含碳酸的总量约在2×10-3mol/L左右,假定有一个pH值小于4.5的水样,当以甲基橙作指示剂并用标准碱溶液进行中和滴定到pH=4.5(指示剂由红色转为黄色)时,所耗用标准碱液的体积数即相当于“无机酸度”。这时水样中的强酸成分全部被碱液中和。向水样加入酚酞指示剂后,继续中和滴定到pH=8.3(指示剂由无色转为红色)时,第二次滴定耗用的标准碱液的体积数相当于“游离二氧化碳酸度”。在这个过程中,由原水样中碳酸所提供的酸度按以下反应被碱液中和:

无机酸度与游离二氧化碳酸度之和合称酚酞酸度。对原水样继续作第三次滴定,达到pH=10.8时所耗用的标准碱液体积数和前两次滴定所耗体积数之和相当于水样的“总酸度”。在这个pH值下,水样中可和滴定碱液相作用的酸性物质全部被中和,但由于中和曲线在此点没有明显突跃,也没有适宜的酸碱指示剂可用于确定终点,所以这项“总酸度”只能是理论性的,一般并不进行这种测定。在第三次滴定中发生的反应为:

另假定有一个pH>10.8的水样,当用标准酸溶液进行中和滴定到pH=10.8时,所耗用的标准酸体积数相当于“苛性碱度”,这时水样中的强碱性成分全部被酸液中和。但由于同上述总酸度不可实测的同样原因,苛性碱度也是无法测定的。向水样加入酚酞指示剂后,继续中和滴定到pH=8.3(指示剂由红色转为无色)时,两次滴定耗用标准酸的体积数之和相当于水样的“酚酞碱度”。在第二次滴定过程中发生如下反应:
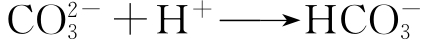
向水样加入甲基橙指示剂后,继续中和滴定到pH值等于4.5(指示剂由黄色转为红色)时,则三次滴定耗用标准酸体积数的总和就相当于“总碱度”或称为“甲基橙碱度”。在第三次滴定过程中发生如下反应:
到此为止,水样中可和滴定酸液相作用的碱性物质全部被中和。各国惯用的酸度和碱度单位不尽一致,应用时要注意区分,并掌握各单位间的转换关系。
按上述,酚酞碱度、甲基橙碱度(即总碱度)是经过采得水样后可实际测定的水质指标。但将对碱度有贡献的苛性碱度(OH-)、碳酸盐碱度(碳酸根离子)和重碳酸盐碱度(碳酸氢根离子)三者从总碱度中区分开来会具有更大实用意义。为此,已提出多种可供使用的方法。这里介绍的是通过实测pH与总碱度Alk值而后求得这三种碱度的平衡计算法。现限定温度为25℃,且以mol[Ca-CO3]/L作碱度单位。由实测pH值首先可求得: