新药Ⅰ期临床试验设计
新药Ⅰ期临床试验的目的是评价新药的耐受性和安全性。当新药从临床前实验转化为Ⅰ期研究(也称为“首次人体”研究)(first in human,FIH)时,此时人们对于人体对这种新药的耐受性和安全性反应还不十分了解,进行初步的临床药理学及人体安全性评估,目的在于了解剂量反应、毒性关系,进行初步的安全性评估,研究人体对新药的耐受性,药效学以及药物在体内的吸收、分布、代谢、排泄等药代动力学,以期推荐Ⅱ期试验初步的给药剂量并有助于Ⅱ期试验的方案设计。
Ⅰ期临床试验受试者一般为健康志愿者,在有些情况下比如肿瘤新药试验,由于抗肿瘤药物所具有的毒性,只能以肿瘤患者作为受试者。在新药Ⅰ期临床试验设计时,必须仔细研究临床前动物实验数据和报告,对新药的作用机制、方式、效价、剂量/浓度-毒性关系、剂量/浓度-效应关系、试验分组、试验人数、剂量递增模式、给药途径、可能产生不良反应的概率及严重程度等进行考虑和评价。在做Ⅰ期试验前不知道人体剂量范围是多少,首先要用动物数据推算出人体的最低剂量,那么到达什么剂量,就会出现毒性呢?什么是人体的安全剂量呢?这里需要介绍三个互相关联的概念:剂量递增的方法、最大耐受剂量(maximum tolerated dose,MTD)、剂量限制性毒性(dose limiting toxicity,DLT)。新药首次在人体上应用(first in human,FIH),探索DLT是FIH的主要目的。人们通过Ⅰ期试验来了解这种新药的耐受性、安全性,确定正确DLT,从而发现最大耐受剂量,对这个药物今后Ⅱ期临床试验选择合适的剂量起到至关重要的作用。DLT有很多影响因素包括药物的种类、作用机制、相关疾病。它是以药物的某些毒性的严重程度来定义DLT,比如在细胞毒性的抗肿瘤药物中,一般把DLT定义为3级的非血液学毒性和4级血液学毒性,但某些2级的毒性,如肾毒性和心脏毒性也可以作为DLT。
在Ⅰ期临床试验中,当受试者对一个低剂量有很好的耐受时,就需要逐步提高试验剂量并最终确定DLT。因为一般来说药物都有量效关系,即给药剂量高了,药物浓度会随之增高,疗效也会提高。这也就是说,一个较高的剂量更能有效地杀死肿瘤细胞,同时药物的毒性也可以杀死正常的细胞,所以剂量越高带来严重毒性的风险也会越大。不管药物的疗效如何,如果毒性太大,超过人体的耐受性,它就无法被接受,那么这种药物也就不能用于临床。Ⅰ期临床试验中,从低剂量到高剂量去寻找DLT,以观察受试者对药物的耐受程度和临床获益。大多数Ⅰ期临床试验都会确定一定数量的剂量梯度供筛选剂量,按照事先所确定的比例递增。表4-6列举了常用剂量递增的方法,包括修正改良Fibonacci法。
表4-6 剂量递增的方法

注:①Penel and Kramar,2012.②Penta,et al.,1979.
为了确定药物的最大耐受剂量(MTD),Dixon和Mood在1948年提出了Up-and-Down设计,在半个世纪后Storer基于这种理念,提出了一种具有可操作性的剂量递增或递减的设计法,即传统3+3设计法(Storer,1989)(表4-7),在这个试验中MTD的定义就是在3+3试验中,同时满足两个条件:①当某个剂量组中≤1/6出现DLT。②邻近高剂量组中≥2/6出现DLT,采用CTCAE V4.0标准来评价药物毒性反应。剂量限制性毒性(DLT)定义可以根据试验的药物不同、适应证不同来制定标准。在肿瘤新药试验中DLT的定义为“4级或以上药物引起的相关血液毒性反应,或者3级或以上药物引起的相关非血液毒性反应,比如心、肝、肾等的毒性反应”。
表4-7 某Ⅰ期临床试验最大耐受剂量设计

注:Gulley J,2016.
3+3设计法具体方案执行如下:每个剂量组最初入组的3名受试者,第1日只安排1名受试者使用研究药物,在确信这位受试者没有出现DLT时,安排同组的其他受试者参加试验。按照顺序依次使用研究药物。为了减少风险,建议不要同时给3名受试者使用同样剂量;在首次、第二次和之后每次给药之间设置足够长的观察期,至少要覆盖预期的峰浓度和(或)峰效应时间。同一日内给药间隔的设置要考虑到可能的急性不良反应发生所需的时间。例如,静脉滴注给药的观察时间应至少为输液的时间(例如,不少于60分钟)。在推进到更高剂量组时,也同样要考虑到不良反应的风险,根据非临床和临床药效学(pharmacodynamic,PD)和药代动力学(pharmacokinetic,PK)数据确定组间的时间间隔。随着免疫疗法的新药开发,免疫疗法不同于化药,每位受试者用药的间隔时间要更长。这里举一个例子,发生于2016年的“大象人”新药试验事件,TNG 1412是一个抗CD28人源化单克隆抗体,由德国TeGenero制药公司研制的新药用于治疗白血病,自身免疫系统疾病如多发性硬化症、风湿性关节炎等。Ⅰ期临床试验在英国伦敦一家医院(Northwick Park Hospital)的Ⅰ期临床试验病房进行。试验计划入组19~34岁的健康志愿者。2006年3月8名健康者自愿参加了试验,其中2人注射安慰剂,其他6人接受研究药物注射。在接受注射几分钟后,6名受试者相继出现了头部肿大,像电影《象人》(Elephant Man)中的主角一样,而且疼痛难忍,甚至出现昏迷以及严重的过敏反应,其中2人出现生命危险,被立即采取抢救措施。这种药物是首次在人体进行试验,此事件迅速引起全球媒体的高度关注,事后分析原因认为这个严重反应主要是由于不可预见的生物学作用所引起的。从新药开发的角度来说,有许多方面值得探讨和改进,但是在试验方法和程序上,之所以6个人同时出现这样的反应,主要是在进行实验的过程中,是一次性的给6个人同时用药,第1个受试者注射2分钟后,再给下一个注射,而不是先给其中一个用药,观察反应以后,再给另外一个受试者用药。如果是这样,一旦一名受试者出现如此严重反应,这个试验可能马上就停止,从而避免更多的受试者受到不必要的损害。
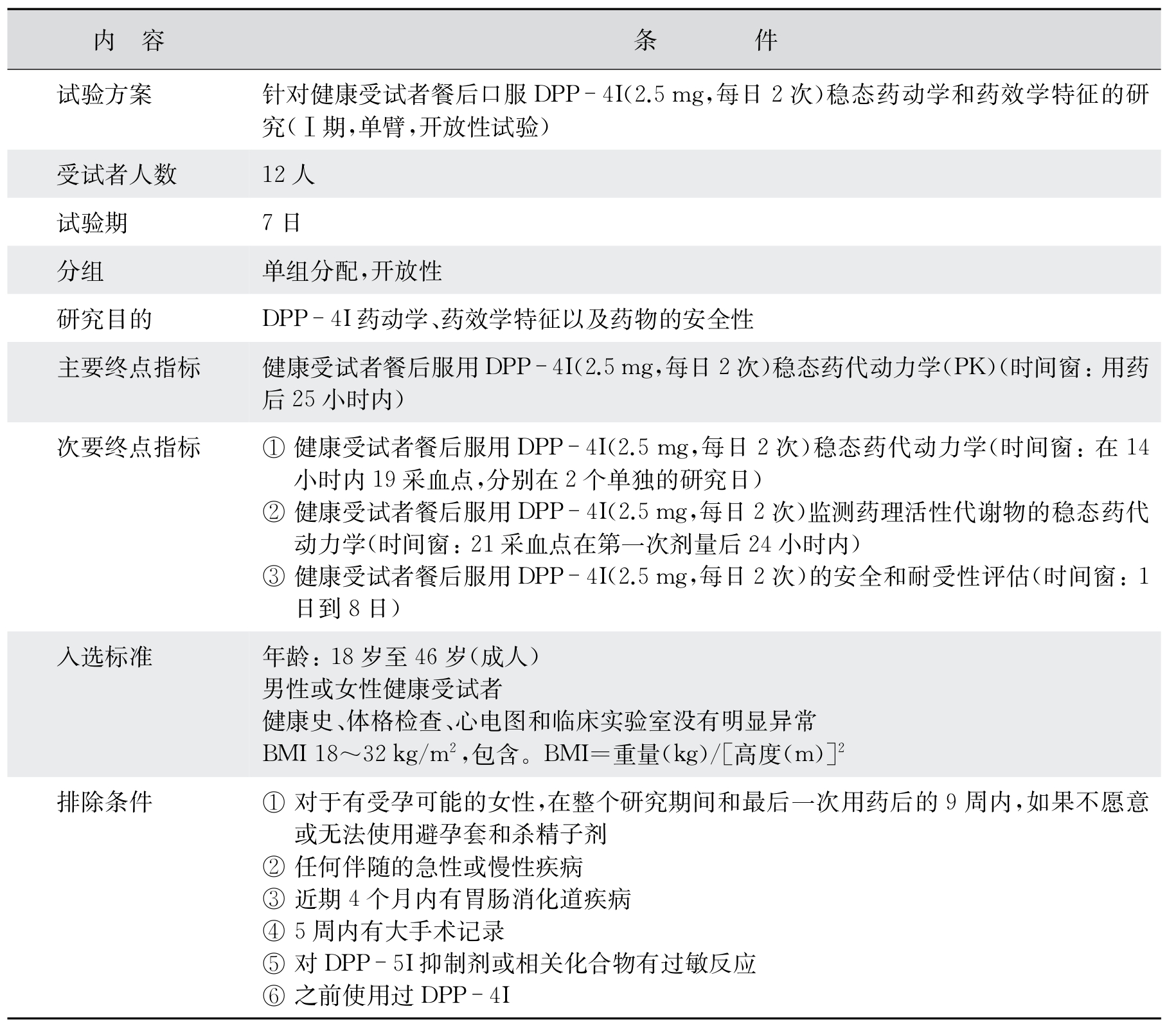
根据FDA指南(Guidance for Industry Diabetes Mellitus:Developing Drugs and Therapeutic Biologics for Treatment and Prevention),对于非胰岛素类的降糖药物,应该在Ⅰ期试验中进行绝对生物利用度、AUC、Cmax、Tmax、T1/2药效学的一些参数检测,这些试验可以在健康受试者中进行。也可以在Ⅱ期试验中的糖尿病受试者中加入药代动力学研究。对于药代动力学的观察和试验设计主要集中在餐后血糖的监测。这样通过药效学和药代动力学的研究可以了解这种药在人体内对新药的耐受性,药效学及对药物在体内的吸收、分布、代谢、排泄等的药代动力学数据,以及了解剂量反应与毒性、进行初步的安全性评估,为制定Ⅱ期试验初步的给药剂量和方案提供相关的数据。新药DPP-4I的Ⅰ期临床试验试验实例如表4-8。DPP-4抑制剂或列汀类药物,是一种通过抑制二肽基肽酶-4来发挥作用的口服抗糖尿病药,用来治疗2型糖尿病。这里以一个DPP-4抑制剂的Ⅰ~Ⅲ期试验方案为例。用药以DPP-4I代替,公司名称省去,方案来源于Clinical Trial.gov。
表4-8 新药DPP-4I试验实例