临床前实验阶段
此阶段包括产品原料药的开发、制程、剂型及动物毒理试验等。药物最终目的是要在患者身上证明有效,但为了安全起见,一定要先在动物身上证明其具有药效,并且安全。为了达到这个目的,有许多实验必须在人体临床试验前完成,才能向药物监管机构申请试验新药(investigational new drug,IND),通过审核批准后再执行临床试验。以下简述IND所需完成的研发项目。
1.化学、制造与监控(chemical manufacture and control,CMC) 主要包含化合物的大量制造、纯度分析、物化性质、安定性试验、剂型设计等。原料药合成工艺研发(process R&D),这是一个不断改进、完善的过程。第一批提供的原料药主要用于毒理研究(100~1 000 g),要求是越快越好,成本不是主要考虑因素。但随着项目的推进,工艺部门会根据需要,设计全新合成路线,开发合理生产工艺来满足Ⅰ~Ⅲ期临床用药与商业化的需求。同理,制剂部门首先也会以最简单的形式给药,完成毒理研究,然后不断完成处方工艺研究,开发出商业化的制剂工艺。
2.药代动力学(pharmacokinetics,PK) 了解药物如何在体内被吸收、分布、代谢、排泄,这些数据可提供未来临床试验给药途径的依据,如口服、注射剂、吸入剂等。
3.安全性药理(safety pharmacology) 证明该化合物针对特定目标疾病具有生物活性,同时评估药物对疗效以外的作用,需进行动物安全性药理试验,以了解可能的副作用,尤其对心血管系统、呼吸系统、中枢神经系统、消化系统等的影响。
4.毒理实验(toxicology) 毒理研究种类较多,包括急性毒性、亚急性毒性、慢性毒性、生殖毒性、致癌性、致突变性等。为了使新药能及早评估是否有疗效,有些毒理实验如致癌性、生殖毒性,可放在临床试验Ⅰ、Ⅱ期同时执行。
5.制剂开发(preparation development) 早期制剂研究并不需要完整的处方开发,所有研究围绕毒理学研究和Ⅰ期临床时方便给药即可,目的是将候选药物尽快推向临床。随着项目推进,给药方式和处方研究越来越全面。比如,有的药胃肠吸收很差,就需要开发为注射剂。有的药在胃酸里会失去活性,就需要开发为肠溶制剂。有的化合物溶解性不好,也可以通过改变制剂来解决这个问题。(https://www.daowen.com)
新药临床试验申请(investigational new drug application,INDA),也就是向药品监督管理部门申请批准新药临床试验,指在原创新药的研发过程中,从动物实验中获得能够安全用于人体的一些基本信息和数据,然后才能申请进入临床试验。申请被批准后,才能进行各期临床试验,一旦证明有效,可以进行新药上市申请,IND才正式结束。获准药品进入人体试验后的IND阶段包括Ⅰ期、Ⅱ期和Ⅲ期临床试验。IND阶段的每个临床试验方案和临床试验总结报告需要及时申报国家药品监督管理部门。在临床试验中出现的药物不良反应和年度报告也需要及时报告。新药上市申请(new drug application,NDA)要提交包括从分子治疗机制开始的所有药品质量、临床前和临床研发数据,目的是要证实药品的安全、有效和优质。NDA一旦生效,其来源的IND随即废止。NDA一直跟随药品上市全程。除了及时提交严重不良反应的报告,在药品上市的每周年需要提交该药品的NDA年度报告。
在临床前阶段,也就是在申报新药临床试验申请之前,需要花费5~10年的时间。①用于研究药物对疾病的作用,在分子水平基础上,研究药物的靶点和作用机制。②药物有效成分的化学合成、提纯、筛选和生产。③用对靶细胞亲和力最大的几个合成分子,在动物体外和动物体内进行药理学、药代动力学和长期或短期毒理学实验,也就是在动物模型上做安全性实验,验证药物的作用机制(mood of action,MOA),同时找出其中一个能够在人体使用的安全起始剂量。
在临床前研究的基础上,对药物的各种质量参数进行评价,把临床前实验数据汇总成总结报告,再加上临床试验方案,就构成了IND的主要申请资料。经过国家药品监督管理部门对提交资料的审查,确认新合成的药物不会对人体构成不必要的风险,就可以开始临床试验。在依次完成Ⅰ期、Ⅱ期、Ⅲ期临床试验后,申办者可以整合所有的数据报告,做新药上市申请。新药上市以后,再做Ⅳ期临床试验,以进一步评价更大人群中用药的药物安全性。
从最初的筛选上万种左右潜在的化合物,到最后有一种新药上市,涉及化学、药理学、毒理学、医学、技术转移、规模扩大、生产质量管理、临床试验伦理、注册资料整理等步骤,是一个非常复杂的系统工程。有学者分析了98家药企10年来所开发的新药,对于那些有能力在10年内开发出8~13种新药的公司,开发1种新药的费用最高可达55亿美元,包括多国多中心试验和药物警戒等安全性的观察(Herper,2013)。所以新药上市是一个高技术、高投资、高风险漫长的过程。这样的过程不仅仅局限于小分子的化学药物,也见之于风险更大、开发难度更复杂、花费更多的大分子(比如抗体及疫苗)生物药的开发。开展人体新药临床试验在药品开发过程中是一个非常重要的里程碑。对药企来说,完成新药临床试验,可能有1种新药上市,同时也标志着该药被国家药品监督管理部门正式纳入监管范围。经过药品监督管理部门的审核批准,这种合成分子才能开始进入临床试验,探索在人体使用的既安全又有效的剂量。经过Ⅰ期、Ⅱ期和Ⅲ期临床试验,分别在健康人群、小批量和大批量患者中试用药物,得到进一步的安全性和足够的有效性依据之后,该药品才能得到药品监督管理部门的审核批准进入市场。图1-1是以美国药品开发的基本模式来显示临床前研究、临床试验和NDA审批三个阶段之间的关系和一些重要的里程碑(FDA,2015)。
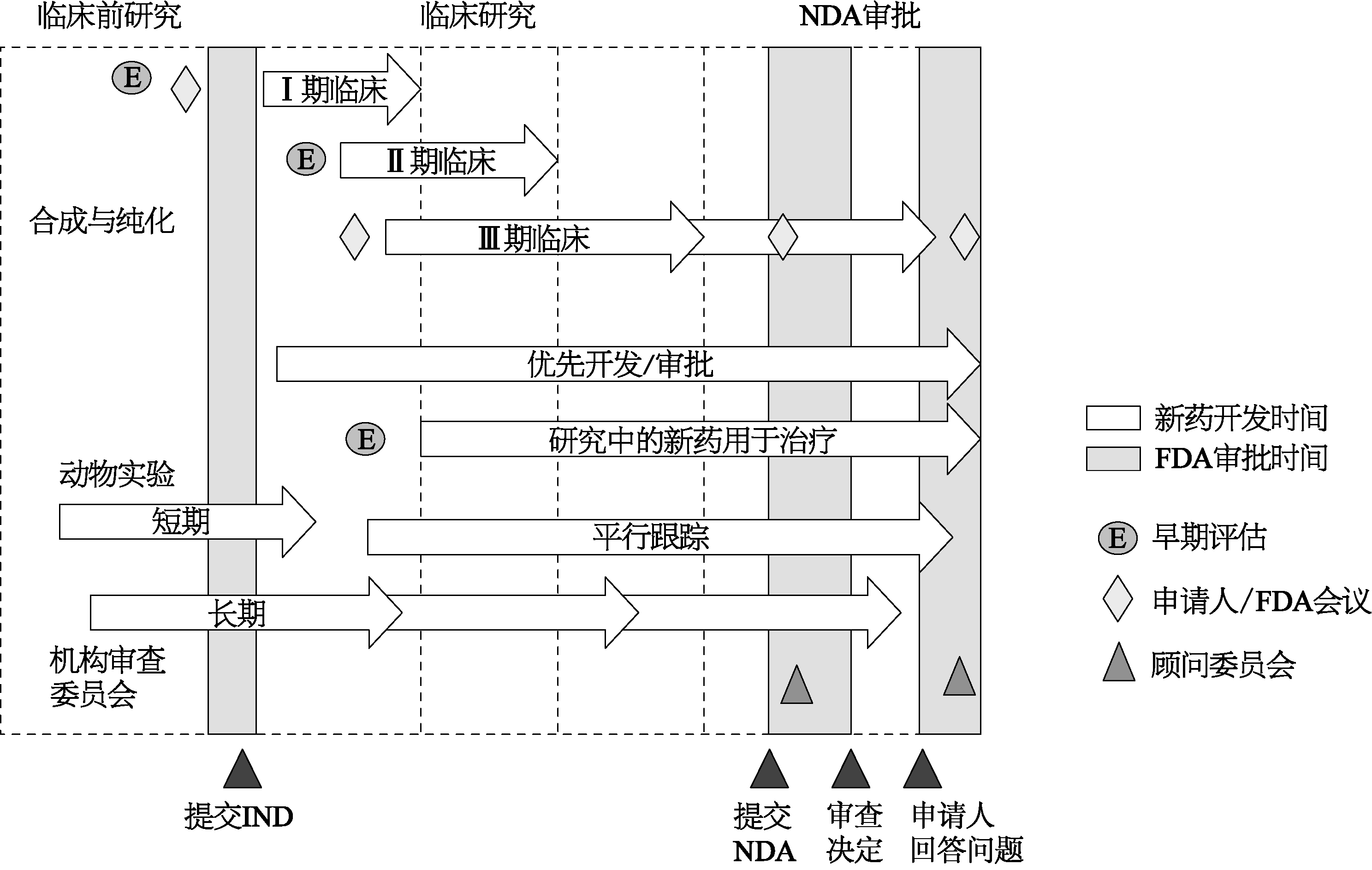
图1-1 美国新药开发过程示意图