药物警戒的操作步骤
药物警戒包括临床试验数据、安全呼叫中心、自发报告和文献检索,每个领域都有可以建立一个单独的系统。根据药品监管政策、法规和指导文件,药物警戒部评估每个严重不良事件和试验药品的因果关系,对综合数据进行系统分析,做获益与风险的安全评估,以快速报告或汇总报告的模式。这在整个产品的生命周期中继续进行。图11-3显示了PV相关的主要活动的流程。
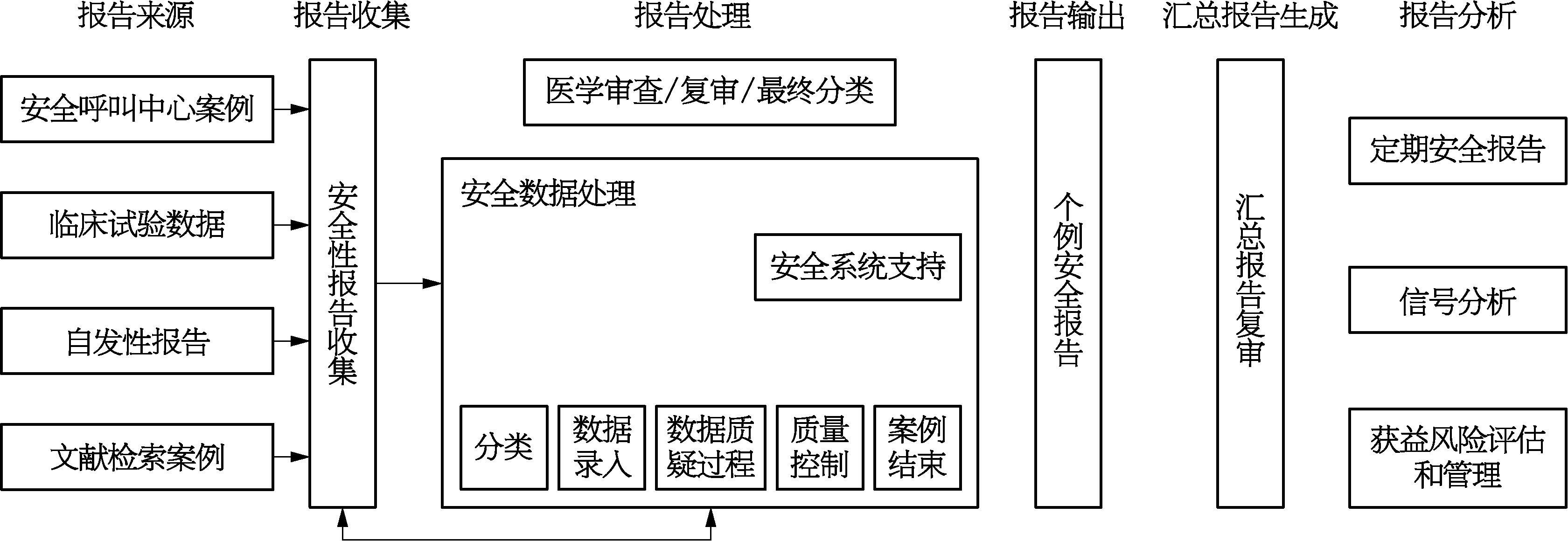
图11-3 药物警戒相关的主要活动
目前严重不良事件的报告与记录通常都采用无纸化。所有的这些报告的处理和审查,都在药物警戒系统之中完成,这个审查的过程,都应该在SOP当中详细地列出来,每一个步骤都清清楚楚的记录,有利于今后的稽查。图11-4描述了这一工作流程。

图11-4 安全数据库严重不良事件案例处理流程图
SAE处理步骤:①当研究者或者监查员发现了一个SAE,那么这个事件就应该立即通报给申办者。②一旦申办者或者CRO收到了SAE报告,就应该立即评估报告是不是符合SAE报告的基本要求。③如果是有效的报告,就要进行进一步检查,是不是有同一事件重复报告。如果不是重复报告,是已经通报过的SAE报告,那么现在这个报告就不是初次报告(initial report),而是随访报告(follow-up report)。④如果报告确认为有效的初次报告,还要检查这个事件是预期的还是非预期的,这个事件和研究药物有无相关性,这个报告是不是符合严重不良反应报告的要求。此外,还要审查这个事件是不是死亡或者是危及生命的事件。如果是死亡病例或者是危及生命的事件,还要在法规规定的时间内处理完成,并且报告到国家药物安全监测中心。⑤然后这个报告要输入到数据库,要撰写该份报告当中的病史描述部分,还要交给药物警戒医师做初步的审查。如果有一些信息不清楚的,还需要和医院进行澄清和确认。所以步骤完成以后还要进行质量监控的审查。⑥同时报告送到国家药物安全监测中心和医院伦理委员会时,还要确认是不是在规定的时限内完成。
在多国多中心实验中,SAE的通报包含两个不同的层次:其一,试验医院一旦发现SAE,除了要按照本国药事法规机关的要求,向本国药事法规机构通报外,还要向申办者通报。其二,各个国家对SAE通报的表格有特别的要求,特别是语言的使用上。申办者可以自己处理这些SAE报告,也可以把这一业务外包给CRO来进行处理。为什么SAE报告除了向药事法规机构通报外,还要向申办者通报呢?从申办者角度来说,在试验结束后,会有一份关于该试验的报告,这份报告不但要反映所研究的新药的确定疗效,还要反映该药物的安全性,因为有必要建立一个安全性数据库(safety database)。据笔者观察,几乎100%的初次SAE报告均存在各种问题,如资料的准确性不够、有些项目未填或错填。对这样的报告进行分析,显然不科学。因此有必要由专人进行SAE不良反应的通报和监测,通常由药企和CRO公司的药物警戒部门来执行。研究者按照方案及申办者的要求,在发现SAE时,填写由申办者提供的SAE表格,在24小时内传真给CRO药物安全部,该部门则由药物安全专员及安全监测医师,按照SOP及安全性管理计划进行审阅,如有需要澄清的问题,会和研究护士及研究者联系。研究护士在澄清这些问题后,再重新把完整的SAE报告传真给CRO,这一过程应在24小时内完成。CRO再把这个SAE报告发给申办者,目的是为了输入安全数据库,以便于将来做安全性数据的统计分析。