文件管理和项目管理工具
项目管理的起始阶段,项目经理需要制定相关文件,同时准备好管理文件的系统。在临床试验行业,有一种说法叫作“没有记录就等于没有发生”。这是一种比较极端的说法,没有记录不是真的等于没有发生。这种说法只是强调在没有记录的情况下,没有证据证明这件事情发生过。所以,在以证据为基础的临床试验行业,往往会将这种情况当成没有发生一样。这说明了在临床试验行业,记录的重要性。而文件是记录的载体,所以临床试验的文件管理至关重要。不管是临床试验的监查、稽查,还是药监部门的检查,都是对临床试验文件进行审核,通过审核文件来还原过程。临床试验的过程通常是无法直接审核的,因为那是已经发生过的事情。有专业人士建议对药品编盲的过程进行录像,但那只是在对临床试验人员高度不信任前提下的产物。临床试验是一个以诚信为基础,以彼此的信任为前提的行业。如果用录像录音的方式来开展临床试验,一方面会涉及受试者隐私权的问题,另一方面也会让临床试验的实施、监查、稽查变得极其困难,所以这种方式不可能成为常规的手段。所以,对临床试验过程的检查,是通过对文件的审核来实现的,这就说明了良好的文件管理的重要性。
项目经理在启动一项临床试验项目以前,首先需要准备的是项目管理的工具。这些项目管理的工具实际上就是一些文件或系统。临床试验的这个行业并不需要很多硬件设备,配有笔记本电脑、打印机、扫描仪、手机等设备即可开展工作。在提供项目管理工具以前,先要准备一个工具箱,可以让工具摆放整齐,便于查找,也不容易漏掉必需的工具,这个工具箱就是临床试验的文件管理系统。
临床试验的文件管理系统可以是传统的纸质文件的管理系统,也可以是电子化文件管理系统。电子化文件管理系统是电脑和网络技术应用于临床试验的结果,也是临床试验随着科技的发展而发展的趋势,但两种系统管理的文件都是一样的。
临床试验的文件管理又分为研究者文件的管理系统和申办方文件的管理系统。现在研究者的文件管理系统大多还是纸质的文件管理系统,也就是一个个的文件夹。申办方的文件常被作为临床试验总文件(trial master file,TMF)。大多数的申办方临床试验总文件都可以采取电子化的管理,但是诸如财务合同、伦理批件、临床试验批件等还是需要纸质文件的。不要认为有扫描件就够了,然后将原件都销毁。我国国家药监局经常需要提供原件资料。就像身份证复印件不能代替身份证一样,原件还是十分重要的。
对于项目的必需文件,以前主要是根据ICH E6 R2[E6(R2)good clinical practice:integrated addendum to ICH E6(R1),guidance for industry]的第八个部分:临床试验的必要文件,提供的目录。ICH E6(R2)的第八部分将临床试验的必要文件分为了三个部分,临床试验启动前的必需文件、临床试验实施过程中的必需文件和临床试验结束后的必需文件。现行的临床试验也是根据这个目录及时地收集各种文件。但是ICH只是给出了必需文件的种类,并没有给出详细的目录。
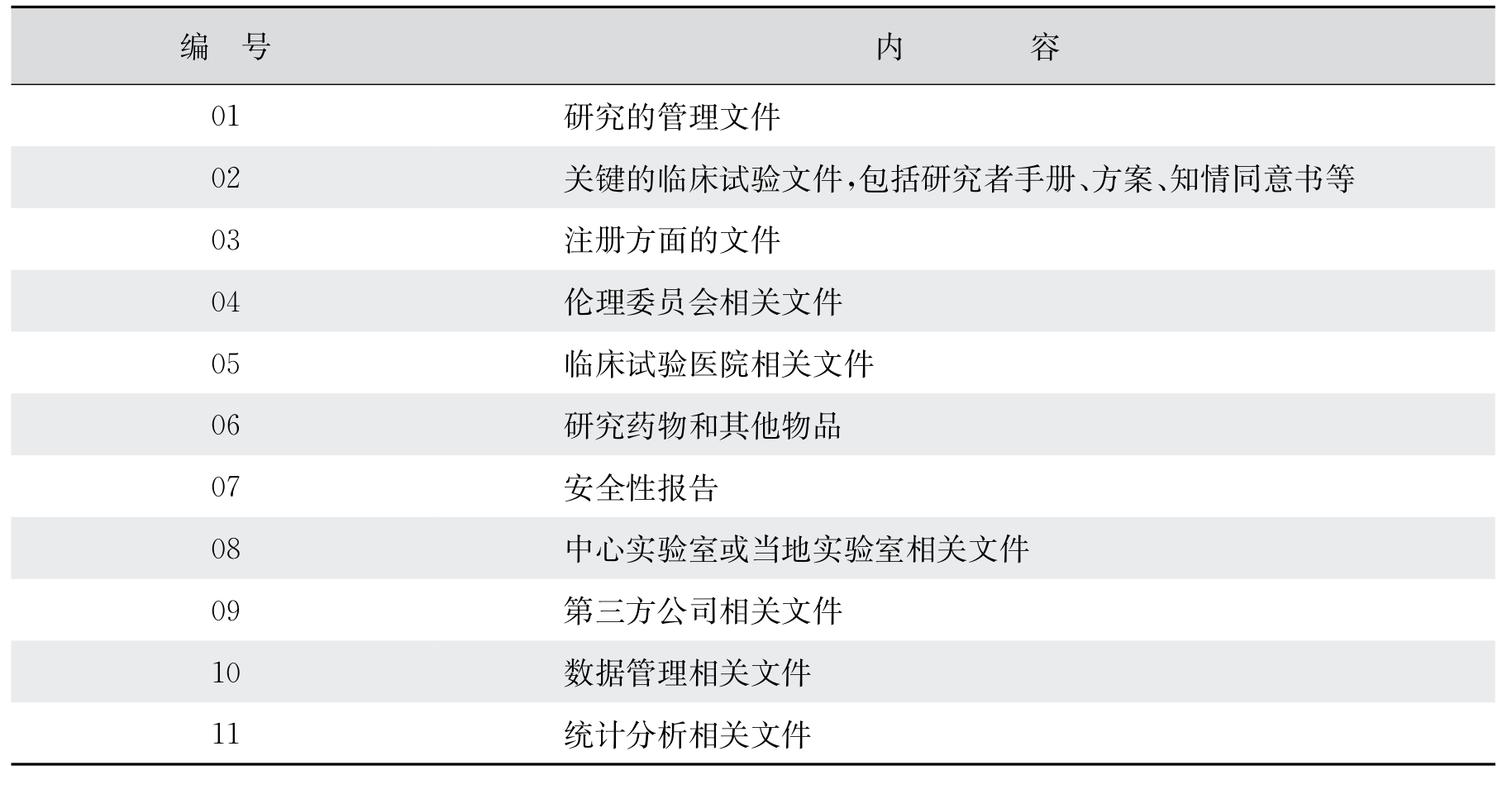
2009年的3月,药物信息协会(Drug Information Association,DIA)召集了300位业内人员组成工作组,讨论并形成了标准化的临床试验总文件目录,对临床试验文件的内容、命名、格式和原数据进行标准化。在之后的时间里,这个标准化的临床试验总文件目录得到了越来越多的申办方或CRO公司的认可。由于对临床试验各个环节进行标准化是一个大的趋势,一旦有公益组织提出一些标准化的程序,大家都是乐于接受的,况且这个标准目录是可以免费使用的。这个目录将临床试验的文件分为从1~11共计11个部分,每个部分又进行细分,形成不同级别的目录和编号,这样相同的文件就会有相同的唯一的编号。表5-2列出了临床试验的文件标准目录,在熟练使用这些目录以后,往往各个文件的编号就被项目管理人员记住了。这样,如果项目经理或监查员跳槽去了其他的公司,由于相同的文件都是相同的编号,就不需要重新再去记忆。
表5-2 临床试验的文件标准目录
一般情况下,公司的标准操作规程(standard operating procedures,SOP)已经提供了临床试验总文件的目录,不需要项目经理做特别的准备。但项目经理在项目启动之初,仍然要核实这个目录,看是否与准备进行的项目有不相符合的地方,确定是否需要进行增减。如果是电子化的项目文件管理系统,就需要对系统进行必要的设定,并对相关人员进行培训和授权。
在制定好临床试验文件管理系统以后,就可以开始制定临床试验项目管理的各种工具。这些工具实际上就是一些文件。表5-3列出了一些常用的临床试验的项目管理工具,不同的公司对项目管理文件有不同的要求。在诸多的项目管理工具中,监查计划已经成为临床试验的必要文件之一。ICH E6 R2的1.64部分规定了监查计划的定义:“监查计划是对临床试验监查的策略、方法、职责和要求进行描述的文件。”在ICH E6 R2的5.18.7中,对监查计划进行了如下的描述:“申办方应该根据项目特点,针对受试者保护和数据的准确和完整方面的风险制定相应的监查计划。监查计划应该对监查的策略、监查各方的责任、不同的监查方法和这些方法的原理描述。监查计划应该强调对关键的数据和程序的监查。需要特别注意那些与常规的临床工作不同,并且需要进行培训的地方。监查计划也要参考相关的政策和程序。”美国FDA在2013年颁布的《基于风险的监查》(A Risk-based Approach to Monitoring)中,对监查计划的内容进行了非常详细的描述,指出制定监查计划应该考虑以下因素:试验设计的复杂性、试验终点的类型、试验人群的复杂性、临床试验医院的地理分布、申办方同研究者的合作经验、是否使用了电子的病例报告表、临床试验药物相对的安全性、临床试验的分期和数据的数量等。同时也给出了监查计划的主要内容,包括:对监查方法的描述、对监查结果的沟通、对不依从情况的管理、对监查质量的保证和监查计划的增补。美国FDA的这一指导原则,对监查计划进行了很详细的说明,可以作为申办方或CRO公司制定监查计划的参考。
表5-3 临床试验的项目管理工具
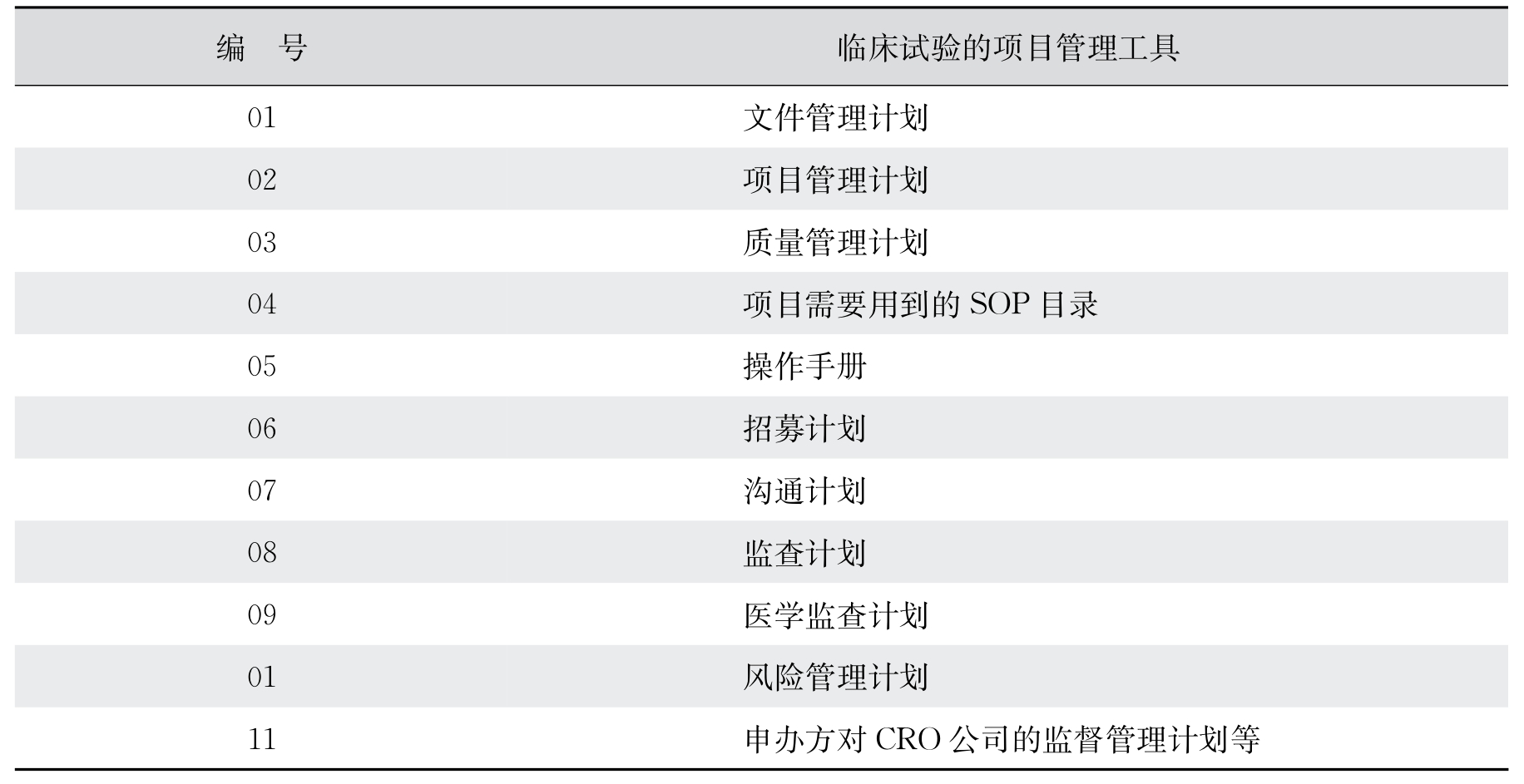
这些工具或文件一般包括但不限于项目文件管理计划、项目管理计划、质量管理计划、本项目需要用到的SOP目录、操作手册、招募计划、沟通计划、监查计划、医学监查计划、风险管理计划、供应商管理计划、申办方对CRO公司的监督管理计划等。也就是说,一位监查员要接手一个临床试验项目,除了要熟悉临床试验方案、知情同意书、研究者手册、中心实验室手册、生物样本的管理、药品管理手册、电子病例报告表填写指南等项目文件以外,还要了解以上的各种计划。不同的公司对于各种计划都有自己的写法。有的公司将某些计划作为项目管理计划的一部分,有的公司将不同的计划单独成文。但不管有多少计划,项目经理需要记住的是,临床试验是在医院由研究者来做的,所有的计划要落实在对在医院第一线的研究者的支持上。如果研究者不按照临床试验的要求完成原始病历,再好的项目管理也毫无用处。同时,根据ICH E6 R2的要求,所有的努力都要最终落实在受试者权益、安全性的保护以及临床试验结果的可靠性上。
对于一个专业的临床试验公司而言,在公司的SOP系统里都可以找到这些工具的模板。在模板的基础上制定各种计划书,对于项目经理而言一般不是一件困难的事情。制定模板的过程是费时费力的,往往需要专门的部门来组织完成。
现实中,有一种误区,认为往往哪个公司制定的文件多,哪个公司的水平就显得高一些,或者说显得更专业一些。但实际上过多而不必要的文件也会增加临床试验的成本,同时也增加了监查员的工作量,让监查员将太多的时间花费在监查以外的地方,这样就舍本逐末了。公司的SOP是临床试验最基本的工具,现在的一些专业的临床试验公司已经建立起庞大的SOP系统,一些监查员连看SOP的时间都没有。所以,太多太过复杂的项目管理工具可能反而会导致一些不依从的问题(non-compliance)。