仿制药品注册管理
(一)对申请人的要求
仿制药申请人应当是药品生产企业,其申请的药品应当与《药品生产许可证》载明的生产范围一致。
(二)对仿制药的要求
仿制药应当与被仿制药具有同样的活性成分、给药途径、剂型、规格和相同的治疗作用。已有多家企业生产的品种,应当参照有关技术指导原则选择被仿制药进行对照研究。
(三)仿制药的申请与审批
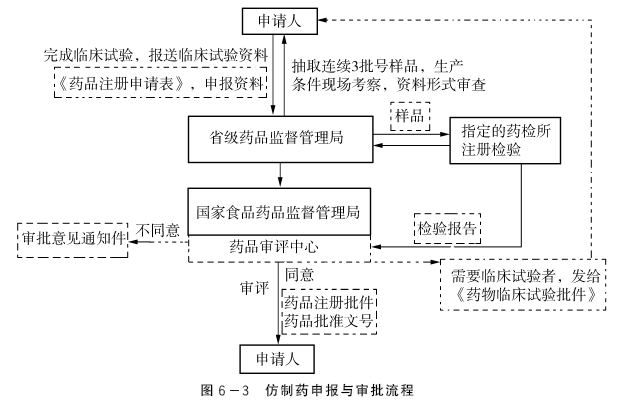
1.申请 申请人填写《药品注册申请表》,向所在地省、自治区、直辖市药品监督管理部门报送有关资料和生产现场检查申请。
2.初审 省局对申报资料进行形式审查,符合要求的,出具药品注册申请受理通知书,并组织进行现场核查,抽取连续生产的3批样品,送药品检验所检验,将审查意见、现场核查报告及申报资料交送药品审批中心。
3.技术审评 药品审批中心对审查意见和申报资料进行全面审评,依据技术审评意见、样品生产现场检查报告和样品检验结果,形成综合意见,连同相关资料报送CFDA。(https://www.daowen.com)
4.审批 CFDA依据综合意见,做出审评决定。符合规定的,发给药品批准文号或者《药物临床实验批件》;不符合规定的,发给《审批意见通知件》。
申请人完成临床试验后,直接向样品审评中心报送临床试验资料。CFDA依据技术意见,发给药品批准文号或者《审批意见通知件》。
仿制药申报与审批流程见图6-3。
(四)仿制药临床试验的有关规定
(1)化学药品注册分类6(已有国家药品标准的原料药或制剂)中的口服固体制剂,应当进行生物等效性试验,一般为18~24例;需要用工艺和标准控制药品质量的,应当进行临床试验,临床试验的病例数至少为100对。
(2)中药、天然药物仿制药视情况需要,进行不少于100对的临床试验。
(3)已有国家药品标准的生物制品和疫苗一般仅需进行Ⅲ期临床,临床试验的病例数至少为300例。
知识链接
美国仿制药品申请上市时的基本要求
在美国,仿制药品申请上市时,除要求遵守《联邦食品、药品和化妆品法案》外,还必须符合《联邦管理法》中的如下规定:
仿制药必须依照FDA《经过医疗等同性评价批准的药品》(Approved Drug Products with Therapeutic Equivalence Evaluations,俗称“橙皮书”)上所列,有FDA选定的参照药品进行对照仿制;其药物活性成分、剂型、规格、给药途径、适应证一定要与所参照药品相同;药品标签、说明书除因专利问题必须删除的部分以及辅料和包装信息等必要的修改外,其他部分必须与所参照创新药品完全一样;必须证明与所参照的创新药具有生物等效性;必须按照《联邦管理法》的规定,遵守GMP要求控制化学生产的工艺过程。
《经治疗等同性评价批准的药品》是FDA出版的在美国所有经安全性和有效性评价,包括处方药和非处方药的被FDA批准的药品名单。此名单列出了各个品牌药品所申报的各项专利以及FDA给予的行政保护期信息并按月更新内容。因为此书的书皮颜色为橙红色,因此俗称“橙皮书”。
(资料来源:中国贸易救济信息网 2007.4.3)