药品不良反应报告和监测管理办法
2010年5月4日,新修订的《药品不良反应报告和监测管理办法》正式颁布,并于2011年7月1日正式施行。国家实行药品不良反应报告制度。药品不良反应报告和监测,是指药品不良反应的发现、报告、评价和控制的过程。药品生产企业(包括进口药品的境外制药厂商)、药品经营企业、医疗机构应当按照规定报告所发现的药品不良反应。国家鼓励公民、法人和其他组织报告药品不良反应。

(一)药品不良反应监测机构及职责
1.国家食品药品监督管理局
(1)负责全国药品不良反应报告和监测工作。
(2)主要职责:
1)与卫生部共同制定药品不良反应报告和监测的管理规定和政策,并监督实施。
2)与卫生部联合组织开展全国范围内影响较大并造成严重后果的药品群体不良事件的调查和处理,并发布相关信息。
3)对已确认发生严重药品不良反应或者药品群体不良事件的药品依法采取紧急控制措施,做出行政处理决定,并向社会公布。
4)通报全国药品不良反应报告和监测情况。
5)组织检查药品生产、经营企业的药品不良反应报告和监测工作的开展情况,并与卫生部联合组织检查医疗机构的药品不良反应报告和监测工作的开展情况。
2.省级食品药品监督管理局
(1)负责本行政区域内药品不良反应报告和监测的管理工作。
(2)主要职责:
1)根据本办法与同级卫生行政部门共同制定本行政区域内药品不良反应报告和监测的管理规定,并监督实施。
2)与同级卫生行政部门联合组织开展本行政区域内发生的影响较大的药品群体不良事件的调查和处理,并发布相关信息。
3)对已确认发生严重药品不良反应或者药品群体不良事件的药品依法采取紧急控制措施,做出行政处理决定,并向社会公布。
4)通报本行政区域内药品不良反应报告和监测情况。
5)组织检查本行政区域内药品生产、经营企业的药品不良反应报告和监测工作的开展情况,并与同级卫生行政部门联合组织检查本行政区域内医疗机构的药品不良反应报告和监测工作的开展情况。
6)组织开展本行政区域内药品不良反应报告和监测的宣传、培训工作。
3.国家药品不良反应监测中心
(1)负责全国药品不良反应报告和监测的技术工作。
(2)主要职责:
1)承担国家药品不良反应报告和监测资料的收集、评价、反馈和上报,以及全国药品不良反应监测信息网络的建设和维护。
2)制定药品不良反应报告和监测的技术标准和规范,对地方各级药品不良反应监测机构进行技术指导。
3)组织开展严重药品不良反应的调查和评价,协助有关部门开展药品群体不良事件的调查。
4)发布药品不良反应警示信息。
5)承担药品不良反应报告和监测的宣传、培训、研究和国际交流工作。
4.省级药品不良反应监测机构
(1)负责本行政区域内的药品不良反应报告和监测的技术工作。
(2)主要职责:
1)承担本行政区域内药品不良反应报告和监测资料的收集、评价、反馈和上报,以及药品不良反应监测信息网络的维护和管理。
2)对设区的市级、县级药品不良反应监测机构进行技术指导。
3)组织开展本行政区域内严重药品不良反应的调查和评价,协助有关部门开展药品群体不良事件的调查。
4)组织开展本行政区域内药品不良反应报告和监测的宣传、培训工作。
5.设区的市级、县级药品不良反应监测机构 负责本行政区域内药品不良反应报告和监测资料的收集、核实、评价、反馈和上报;开展本行政区域内严重药品不良反应的调查和评价;协助有关部门开展药品群体不良事件的调查;承担药品不良反应报告和监测的宣传、培训等工作。
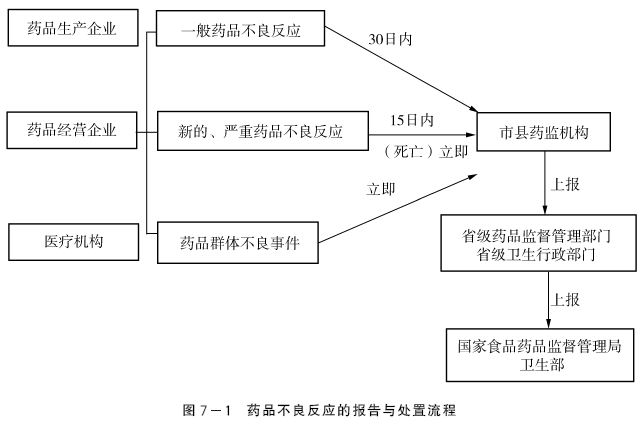
(二)药品不良反应的报告与处置
1.个例药品不良反应
(1)药品生产、经营企业和医疗机构应当主动收集药品不良反应,获知或者发现药品不良反应后应当详细记录、分析和处理,填写《药品不良反应/事件报告表》并报告。
(2)新药监测期内的国产药品应当报告该药品的所有不良反应;其他国产药品,报告新的和严重的不良反应。进口药品自首次获准进口之日起5年内,报告该进口药品的所有不良反应;满5年的,报告新的和严重的不良反应。
(3)药品生产、经营企业和医疗机构发现或者获知新的、严重的药品不良反应应当在15日内报告,其中死亡病例须立即报告;其他药品不良反应应当在30日内报告。有随访信息的,应当及时报告。
(4)药品生产企业应当对获知的死亡病例进行调查,详细了解死亡病例的基本信息、药品使用情况、不良反应发生及诊治情况等,并在15日内完成调查报告,报药品生产企业所在地的省级药品不良反应监测机构。
(5)个人发现新的或者严重的药品不良反应,可以向经治医师报告,也可以向药品生产、经营企业或者当地的药品不良反应监测机构报告,必要时提供相关的病历资料。
2.药品群体不良事件
(1)药品生产、经营企业和医疗机构获知或者发现药品群体不良事件后,应当立即通过电话或者传真等方式报所在地的县级药品监督管理部门、卫生行政部门和药品不良反应监测机构,必要时可以越级报告;同时填写《药品群体不良事件基本信息表》,对每一病例还应当及时填写《药品不良反应/事件报告表》,通过国家药品不良反应监测信息网络报告。
(2)设区的市级、县级药品监督管理部门获知药品群体不良事件后,应当立即与同级卫生行政部门联合组织开展现场调查,并及时将调查结果逐级报至省级药品监督管理部门和卫生行政部门。
对全国范围内影响较大并造成严重后果的药品群体不良事件,国家食品药品监督管理局应当与卫生部联合开展相关调查工作。
(3)药品生产企业获知药品群体不良事件后应当立即开展调查,详细了解药品群体不良事件的发生、药品使用、患者诊治以及药品生产、储存、流通、既往类似不良事件等情况,在7日内完成调查报告,报所在地省级药品监督管理部门和药品不良反应监测机构;同时迅速开展自查,分析事件发生的原因,必要时应当暂停生产、销售、使用和召回相关药品,并报所在地省级药品监督管理部门。
(4)药品经营企业发现药品群体不良事件应当立即告知药品生产企业,同时迅速开展自查,必要时应当暂停药品的销售,并协助药品生产企业采取相关控制措施。
(5)医疗机构发现药品群体不良事件后应当积极救治患者,迅速开展临床调查,分析事件发生的原因,必要时可采取暂停药品的使用等紧急措施。
(6)药品监督管理部门可以采取暂停生产、销售、使用或者召回药品等控制措施。卫生行政部门应当采取措施积极组织救治患者。
3.境外发生的严重药品不良反应
(1)进口药品和国产药品在境外发生的严重药品不良反应(包括自发报告系统收集的、上市后临床研究发现的、文献报道的),药品生产企业应当填写《境外发生的药品不良反应/事件报告表》,自获知之日起30日内报送国家药品不良反应监测中心。国家药品不良反应监测中心要求提供原始报表及相关信息的,药品生产企业应当在5日内提交。
(2)进口药品和国产药品在境外因药品不良反应被暂停销售、使用或者撤市的,药品生产企业应当在获知后24小时内书面报国家食品药品监督管理局和国家药品不良反应监测中心。
4. 定期安全性更新报告
(1)设立新药监测期的国产药品,应当自取得批准证明文件之日起每满1年提交一次定期安全性更新报告,直至首次再注册,之后每5年报告一次;其他国产药品,每5年报告一次。
(2)国产药品的定期安全性更新报告向药品生产企业所在地省级药品不良反应监测机构提交。进口药品(包括进口分包装药品)的定期安全性更新报告向国家药品不良反应监测中心提交。
药品不良反应的报告与处置流程见图7-1。
(三)药品重点监测
(1)药品生产企业应当经常考察本企业生产药品的安全性,对新药监测期内的药品和首次进口5年内的药品,应当开展重点监测,并按要求对监测数据进行汇总、分析、评价和报告;对本企业生产的其他药品,应当根据安全性情况主动开展重点监测。
(2)省级以上药品监督管理部门根据药品临床使用和不良反应监测情况,可以要求药品生产企业对特定药品进行重点监测;必要时,也可以直接组织药品不良反应监测机构、医疗机构和科研单位开展药品重点监测。
(3)省级以上药品不良反应监测机构负责对药品生产企业开展的重点监测进行监督、检查,并对监测报告进行技术评价。
(4)省级以上药品监督管理部门可以联合同级卫生行政部门指定医疗机构作为监测点,承担药品重点监测工作。
知识链接
“尼美舒利”修改说明书
2011年5月,国家食品药品监督管理局下发通知,决定采取进一步措施加强尼美舒利口服制剂使用管理,内容包括:禁止用于12岁以下儿童;作为抗炎镇痛的二线用药,只能在至少一种其他非甾体抗炎药治疗失败的情况下使用;适应证限于慢性关节炎(如骨关节炎等)的疼痛、手术和急性创伤后的疼痛、原发性痛经的症状治疗;最大单次剂量不超过100毫克,疗程不能超过15天,并应依据临床实际情况采用最小的有效剂量、最短的疗程,以减少药品不良反应的发生。通知要求,各地药品监管部门严格依法监督辖区内相关药品生产企业尽快按照《尼美舒利口服制剂说明书修订要求》修订说明书和标签,并将修订内容及时通知相关医疗机构、药品经营企业等单位。要求相关药品生产企业主动跟踪尼美舒利口服制剂临床应用的安全性情况,按规定收集药品不良反应并及时报告。
(四)评价与控制
(1)药品生产企业应当对收集到的药品不良反应报告和监测资料进行分析、评价,并主动开展药品安全性研究。
(2)省级药品不良反应监测机构应当每季度对收到的药品不良反应报告进行综合分析,提取需要关注的安全性信息,并进行评价,提出风险管理建议,及时报省级药品监督管理部门、卫生行政部门和国家药品不良反应监测中心。
省级药品监督管理部门根据分析评价结果,可以采取暂停生产、销售、使用和召回药品等措施,并监督检查,同时将采取的措施通报同级卫生行政部门。

(3)国家药品不良反应监测中心应当每季度对收到的严重药品不良反应报告进行综合分析,提取需要关注的安全性信息,并进行评价,提出风险管理建议,及时报国家食品药品监督管理局和卫生部。
国家食品药品监督管理局根据药品分析评价结果,可以要求企业开展药品安全性、有效性相关研究。必要时,应当采取责令修改药品说明书,暂停生产、销售、使用和召回药品等措施,对不良反应大的药品,应当撤销药品批准证明文件,并将有关措施及时通报卫生部。
(五)法律责任
(1)药品生产企业有下列情形之一的,由所在地药品监督管理部门给予警告,责令限期改正,可以并处5000元以上3万元以下的罚款:
1)未按照规定建立药品不良反应报告和监测管理制度,或者无专门机构、专职人员负责本单位药品不良反应报告和监测工作的。
2)未建立和保存药品不良反应监测档案的。
3)未按照要求开展药品不良反应或者群体不良事件报告、调查、评价和处理的。
4)未按照要求提交定期安全性更新报告的。
5)未按照要求开展重点监测的。
6)不配合严重药品不良反应或者群体不良事件相关调查工作的。
7)其他违反本办法规定的。
药品生产企业有前款规定第4)项、第5)项情形之一的,按照《药品注册管理办法》的规定对相应药品不予再注册。
(2)药品经营企业有下列情形之一的,由所在地药品监督管理部门给予警告,责令限期改正;逾期不改的,处3万元以下的罚款:
1)无专职或者兼职人员负责本单位药品不良反应监测工作的。
2)未按照要求开展药品不良反应或者群体不良事件报告、调查、评价和处理的。
3)不配合严重药品不良反应或者群体不良事件相关调查工作的。
(3)医疗机构有下列情形之一的,由所在地卫生行政部门给予警告,责令限期改正;逾期不改的,处3万元以下的罚款。情节严重并造成严重后果的,由所在地卫生行政部门对相关责任人给予行政处分:
1)无专职或者兼职人员负责本单位药品不良反应监测工作的。
2)未按照要求开展药品不良反应或者群体不良事件报告、调查、评价和处理的。
3)不配合严重药品不良反应和群体不良事件相关调查工作的。
药品监督管理部门发现医疗机构有前款规定行为之一的,应当移交同级卫生行政部门处理。卫生行政部门对医疗机构做出行政处罚决定的,应当及时通报同级药品监督管理部门。
(4)各级药品监督管理部门、卫生行政部门和药品不良反应监测机构及其有关工作人员在药品不良反应报告和监测管理工作中违反本办法,造成严重后果的,依照有关规定给予行政处分。
(5)药品生产、经营企业和医疗机构违反相关规定,给药品使用者造成损害的,依法承担赔偿责任。
案例分析
感冒清片(胶囊)引起的不良反应案
感冒清片(胶囊)为中西药复方制剂,由对乙酰氨基酚、马来酸氯苯那敏、盐酸吗啉胍3种化药成分及南板蓝根、大青叶、金盏银盘、岗梅、山芝麻、穿心莲叶6味中药组方而成。在临床应用过程中,常与含有相同成分或功效类似的药品联合使用,造成组方成分超剂量使用或引起毒性协同作用,可能导致严重不良反应风险增加。
典型病例1:患者男,44岁,因患感冒服用感冒清片,一次4片,一日3次。连续用药3天后患者出现肉眼血尿及尿频、尿痛等尿路刺激征,即入院就诊。查尿常规,镜检见红细胞满视野。嘱立即停用感冒清片,给予对症治疗后,患者血尿及尿路刺激征缓解,好转出院。
典型病例2:患者男,65岁,因“斜颈伴肩部肌肉痉挛”就诊。患者因感冒自行服用感冒灵冲剂(颗粒)及感冒清片,次晨自感颈部不适,头颈歪斜,肩部肌肉痉挛,遂就诊。经问诊,患者发病前同时服用感冒灵冲剂及感冒清片,考虑为药物所致,门诊给予停药,盐酸苯海索口服治疗。两日后患者症状消失,痊愈。
分析:两个案例是否均定性为药品不良反应?我们在日常用药过程中如何了解可能存在的用药风险,从而避免或减少不良反应的发生?