4.4.4 PCR技术的拓展及其应用
1.逆转录PCR
RT-PCR是一种从细胞RNA(mRNA)中高效、灵敏地扩增cDNA序列的方法。顾名思义,它由两大步骤组成,首先是逆转录(reverse transcription,RT),然后是PCR扩增反应。其基本操作过程为提取组织或细胞中的总RNA,以其中的mRNA为模板,以Oligo(dT)(图4-8(a))或随机引物(图4-8(b))为引物,利用逆转录酶逆转录成cDNA,再以cDNA为模板进行PCR扩增,而获得目的基因片段(图4-8)。
2.反向PCR
常规的PCR技术是用于扩增两段已知序列之间的DNA片段,而对于已知序列侧翼的未知DNA序列的扩增,则毫无办法。但是,在很多时候,我们又非常渴望了解某些特征性遗传标记侧翼的序列,则可采用反向PCR技术(inverse polymerase chain reaction,IPCR)。由于IPCR是用于扩增已知序列侧翼的DNA片段,因此其一对引物尽管与常规PCR引物一样与已知序列互补,但方向是相反的(常规PCR引物的方向是相对的)。用这样一对引物进行常规PCR扩增,是无法得到足够的产物的,因为其每个引物只能对各自的模板线性扩增,无法进行指数式增长。所以对用IPCR进行扩增的DNA模板必须先经过酶切,然后连接环化,使其引物方向成为相对的,故IPCR步骤主要包括酶切、自身连接环化、PCR扩增、直接序列分析或克隆后再测序(图4-9)。
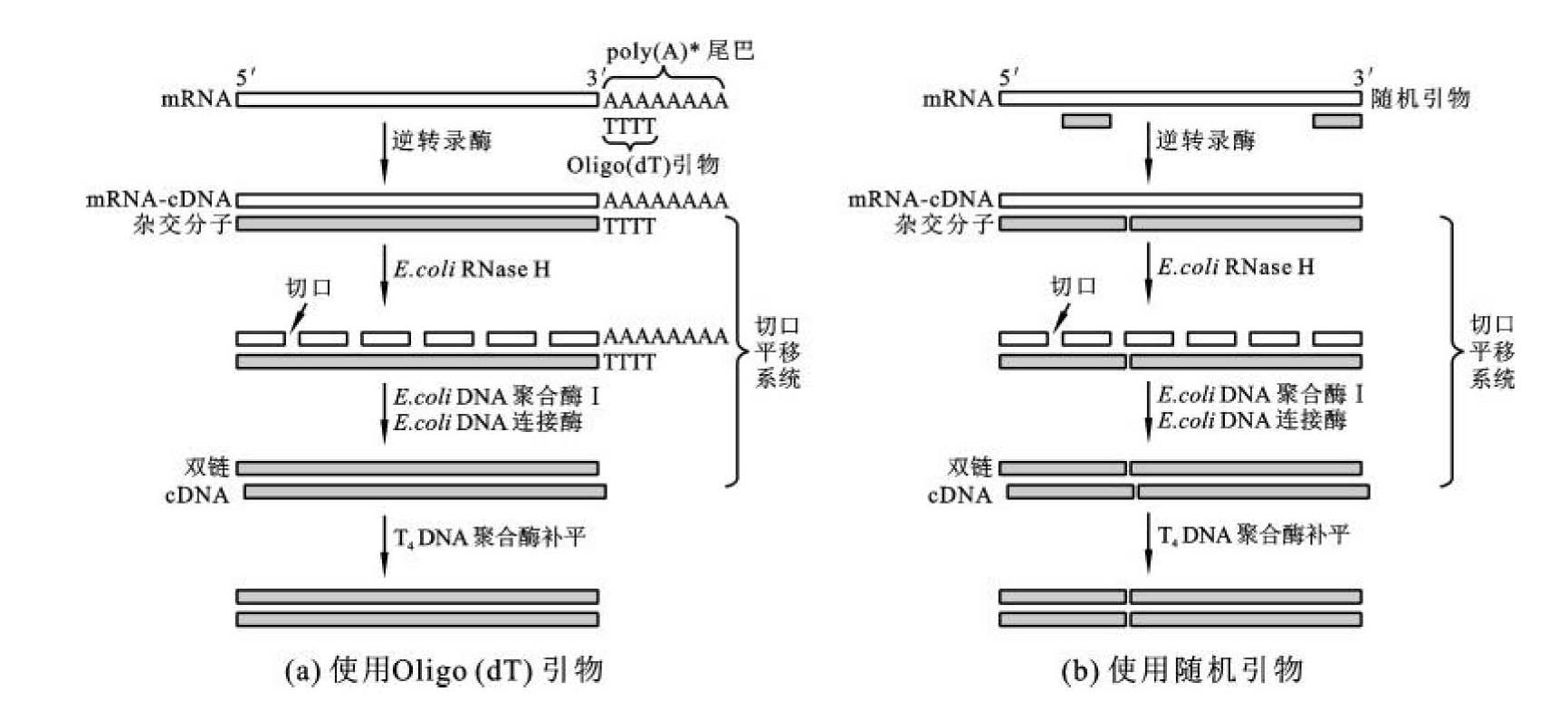
图4-8 使用Oligo(dT)引物或随机引物时的cDNA合成
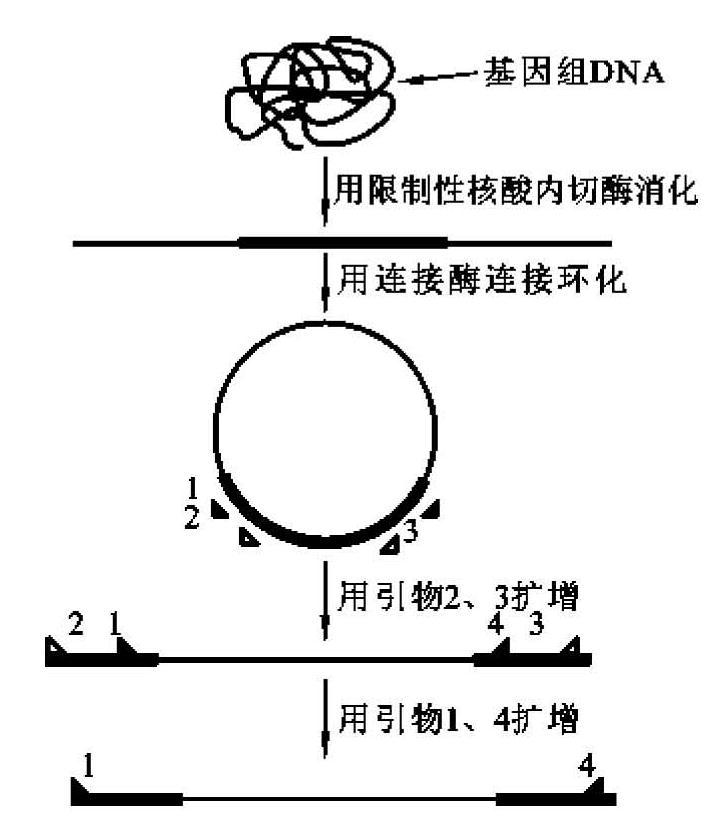
图4-9 反向PCR原理示意图
3.锚定PCR
在实际工作中常仅知道一小段基因序列,难以利用常规PCR技术扩增得到邻近的片段。随着PCR技术的发展,出现了一些根据一小段序列信息快速扩增已知序列相邻片段的技术,其中之一就是锚定PCR(anchored PCR)或锚式PCR(anchor PCR)技术,也称为单侧特异引物PCR(single-specific sequence primer PCR,SSPPCR)。如扩增已知序列3'端的基因,依据已知序列设计上游引物(PS1与PS2),以成熟mRNA 3'端的poly(A)尾巴设计一段Oligo(dT)作为锚定引物,以cDNA为模板通过锚定PCR扩增已知序列3'端的基因(图4-10(a))。这种以cDNA为模板的锚定PCR技术称为互补锚定PCR(complementary anchored PCR)。采用互补锚定PCR技术还可以扩增已知序列的5'端的基因(图4-10(b))。
4.简并PCR
如果要扩增的基因序列不清楚,只知道部分氨基酸序列,例如用双向电泳技术鉴定到一个新的蛋白质,通过测序获得该蛋白质的末端氨基酸序列,要扩增该基因,就要由氨基酸序列推测其核苷酸序列,并以可能的序列设计多条引物进行PCR扩增。如果已知某段氨基酸序列在多种生物中是高度保守区域,要想扩增出这段区域以进行研究,也需要根据氨基酸序列设计引物进行PCR扩增。但编码氨基酸的密码子存在简并性,往往很难准确地确定核苷酸序列,从而给引物设计带来很大的困难。因此,根据密码子简并性,设计出了包含多条核苷酸序列的引物库,利用混合的引物库进行PCR,就形成了一套简并PCR的技术方法。该方法的原理是基于氨基酸序列设计两组带有一定简并性的引物库,从不同生物物种中扩增出未知核苷酸序列的基因。简并引物库是由一组引物构成的,这些引物有很多相同碱基,在序列的好几个位置也有很多不同的碱基,只有这样才会和多种同源序列发生退火,以实现PCR扩增。简并PCR与一般PCR的不同之处在于,一般PCR中的引物是用给定的核苷酸序列设计的两条特定的引物,而在简并PCR中用的是由多条不同核苷酸序列组成的混合引物库,是由一组引物构成的,这些引物有很多相同碱基,在序列的好几个位置也有很多不同的碱基,只有这样,才会和多种同源序列发生退火,以实现PCR扩增。例如“Ala Asn Ile Lys Met”的引物库简并度为4×2× 3×2×1=48(编码简并度:Ala=4,Asn=2,Ile=3,Lys=2,Met=1),具体如下:
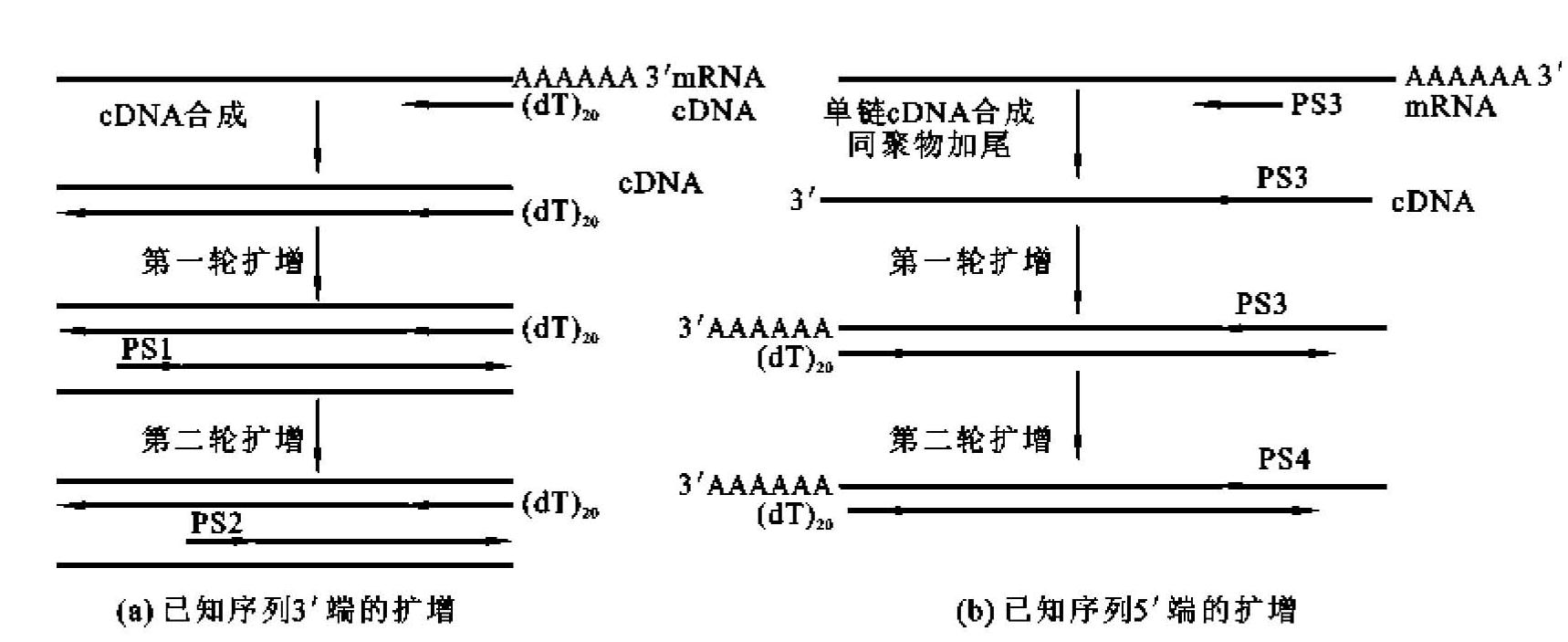
图4-10 用锚定PCR扩增已知序列的上下游基因
Ala Asn Ile Lys Met
5'GCN AAY UAY AAR AUG 3'
其中,N代表A、G、C或T,Y代表T或C,R代表G或A。在引物中有越多的Y、R或N,引物的简并程度就越大。
5.荧光定量PCR
(1)荧光定量PCR的原理:定量PCR技术是利用PCR来测量样品中DNA或RNA的原始模板拷贝数的PCR技术。荧光定量PCR技术则是通过荧光染料或荧光探针,对PCR产物进行标记跟踪,利用荧光信号实时在线监控反应过程。随着PCR的进行,PCR产物不断累积,荧光信号强度也等比例增加,且被激发的荧光强度与扩增产物成正比,每经过一个循环,收集一个荧光强度信号,通过收集到的荧光强度变化监测产物量的变化,结合相应的软件可以对产物进行分析,从而得到一条荧光扩增曲线,计算待测样品的初始模板。荧光定量PCR技术是一次DNA定量技术的飞跃。运用该项技术,可以对DNA、RNA样品进行定量和定性分析。
①SYBR GreenⅠ荧光染料技术原理:SYBR GreenⅠ是一种结合于所有dsDNA双螺旋小沟区域激发一定荧光的染料。SYBR GreenⅠ荧光染料技术原理如图4-11所示,SYBR GreenⅠ只与双链DNA结合才能发出荧光,而不掺入双链中的SYBR染料分子不会发射任何荧光信号。在PCR体系中,加入过量SYBR荧光染料,从而保证荧光信号的增加与PCR产物的增加完全同步。荧光信号强度与双链DNA分子数成正比,随着扩增产物增加而增加,荧光信号的强度代表了反应体系中双链DNA分子的数量。
②TaqMan探针技术原理:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个荧光报告基团(reporter)和一个荧光淬灭基团(quencher)。探针完整时,报告基团发射的荧光信号被淬灭基团吸收,PCR仪检测不到荧光信号。扩增时,Taq DNA聚合酶的5'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的增强与PCR产物的形成完全同步(图4-12)。随着扩增循环数的增加,释放出来的荧光基团不断积累,因此荧光强度与扩增产物的数量成正比,这也是定量的基础所在。相对于染料法,TaqMan探针法具有更高的特异性和准确性。
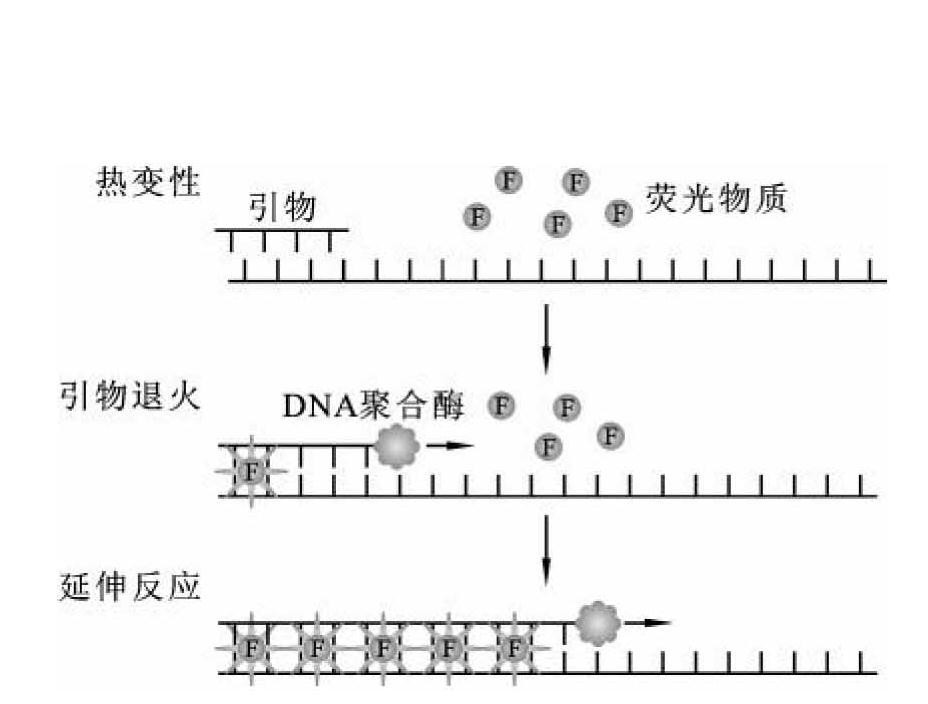
图4-11 SYBR Green Ⅰ工作原理
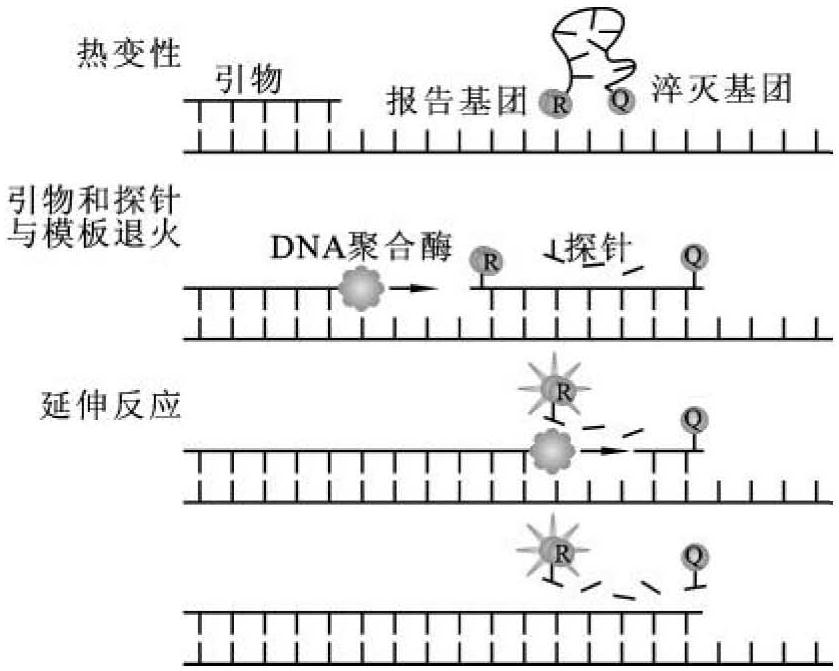
图4-12 TaqMan探针技术原理
③Beacon探针技术原理:该技术加入的荧光探针是环状的寡核苷酸探针,由茎部和环部组成,两端分别标记荧光报告基团和荧光淬灭基团,在无靶序列的情况下,探针始终是环状,报告基团的荧光被淬灭基团淬灭,使荧光检测仪检测不到荧光信号;而在有靶序列时,即在PCR的退火阶段,探针与靶序列结合,使荧光报告基团和淬灭基团分开,这样荧光仪可以检测到荧光信号,荧光信号的强弱代表了靶序列的多少(图4-13)。
④Lightcycler技术原理:该技术以Roche(罗氏)公司为代表。两条直线型寡核苷酸探针,其中一条(D杂交探针)的3'端标记供体(donor)荧光基团,另一条(A杂交探针)的5'端标记受体(acceptor)荧光基团,在无靶序列的情况下,彼此分开,无法进行能量的传递,这样荧光仪不能检测到荧光信号;而当有靶序列时,即在PCR的退火阶段,两条探针与靶序列结合,由于两探针设计时可与模板同一条链相邻的序列杂交,杂交时供体荧光基团和受体荧光基团便紧密相邻,使得两条探针上的荧光基团可以进行能量的传递,这样荧光仪就可以检测到荧光信号,因此可以进行PCR定量分析(图4-14)。该方法由于两个探针结合于模板上而影响扩增效率。同时还需要合成两个较长的探针,因此合成成本相对较高。
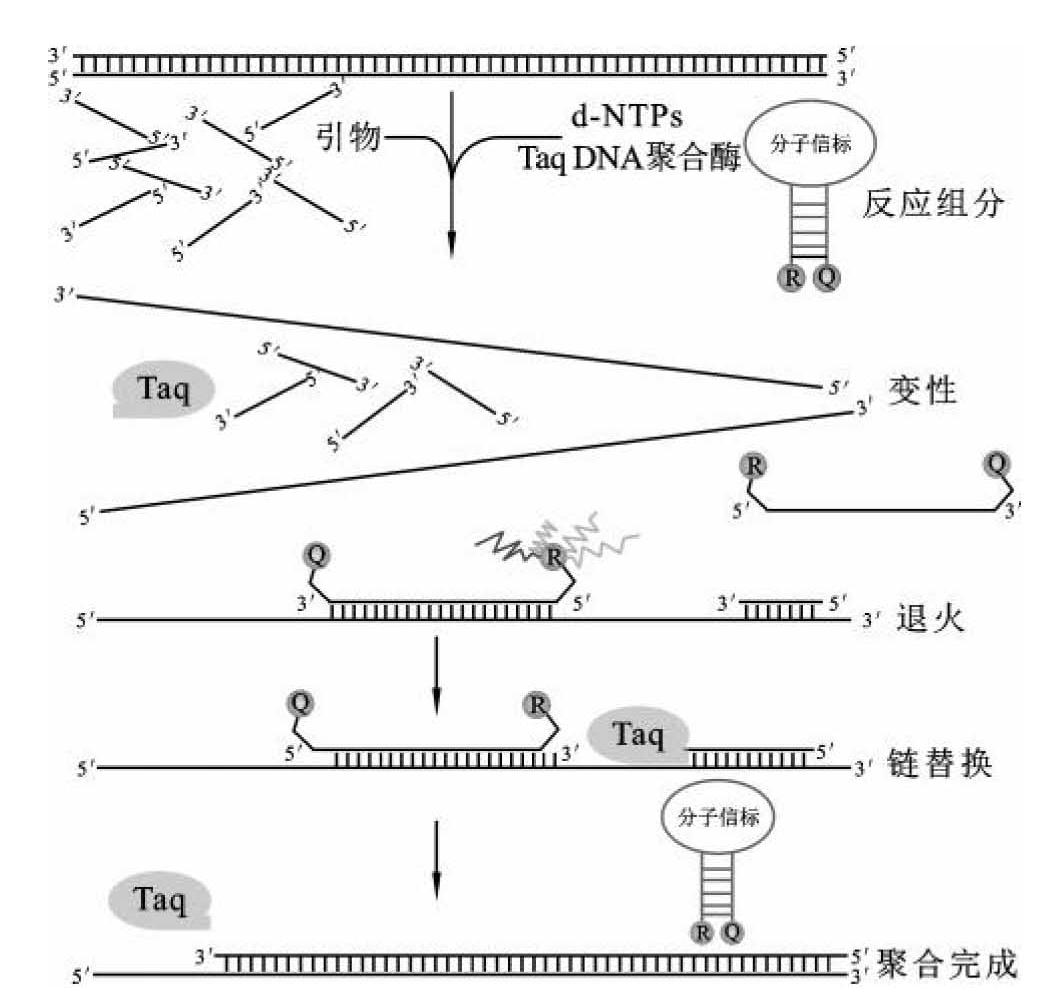
图4-13 Beacon探针技术原理
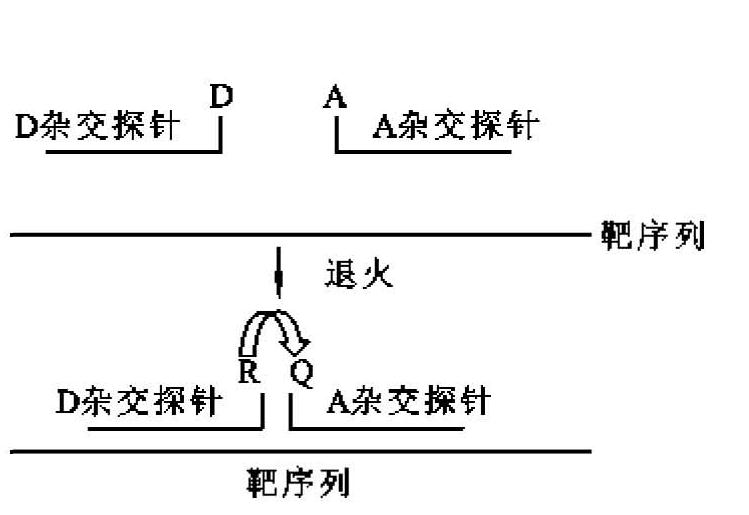
图4-14 Lightcycler技术原理
(2)荧光定量PCR的应用。
①基因表达定量分析:如同一基因在不同组织中的表达差异、同一基因在不同药物处理后的表达差异、转基因食品的检测。过去最常用的高通量筛选基因表达差异的技术是cDNA芯片和差异显示,但这两种技术的缺点是只能定性而非完整意义上的定量分析。实时PCR技术和高通道实时PCR仪的出现,无疑为这种检测提供了极大的方便。
②点突变分析和等位基因分析:用不同的荧光报告基团标记TaqMan探针,然后对等位基因进行荧光定量PCR检测。如果一种荧光信号明显强于另一种荧光信号,则表明它是纯合子(等位基因相同);如果两种荧光信号都明显增强,则表明它是杂合子(等位基因不相同)。如分子信标(molecular beacon)就是专门为这种技术设计的探针。
③单核苷酸多态性的分析:检测单核苷酸多态性对于研究个体对不同疾病的易感性或者个体对特定药物的不同反应有着重要的意义。因分子信标结构的巧妙性,一旦SNP的序列信息是已知的,采用这种技术进行高通量的SNP检测将会变得简单而准确。
④DNA甲基化检测:DNA甲基化同人类的许多疾病有关,特别是癌症,Laird报道了一种被称为Methylight的技术,在扩增之前先处理DNA,使得未甲基化的胞嘧啶变成尿嘧 啶,而甲基化的胞嘧啶不受影响,用特异性的引物和TaqMan探针来区分甲基化和非甲基化的DNA,这种方法不仅方便而且灵敏度更高。
⑤对传染性疾病进行定量定性分析:这在我国应用比较广泛。许多生产临床PCR试剂的厂商已经陆续推出了一系列的诊断试剂,如肝炎系列、性病系列、肿瘤系列等。