13.2.1 定点诱变
随着分子生物学技术的发展,特别是基因克隆技术的应用,分离并研究单基因的结构与功能已成为一种常规的工作。与此相适应,基因诱变技术也有了极大的发展。现在,不仅能够对多细胞或是有机体进行诱变处理,并从成千上万突变群体中筛选出期望的突变体,而且能在体外试管中通过碱基取代、插入或缺失使基因DNA序列中任何一个特定的碱基发生改变。这种体外特异性改变某个碱基的技术,叫做定点诱变(site-directed mutagenesis)。它具有简单易行、重复性高等优点,现已发展成为基因操作的一种技术。这种技术不仅适用于基因结构与功能的研究,还可通过改变基因的密码子来改造天然蛋白质。目前已发展的定点诱变方法主要有M13寡核苷酸诱变、PCR诱变、盒式诱变及随机诱变等,下面将逐一予以讨论。
1.M13寡核苷酸诱变
1)原理
寡核苷酸定点诱变技术所依据的原理是按照体外重组DNA技术,将待诱变的目的基因插入M13噬菌体上,制备此种含有目的基因的M13单链DNA,即正链DNA。再使用化学合成的含有突变碱基的寡核苷酸短片段作为引物,启动单链DNA分子进行复制,随后这段寡核苷酸引物便成为新合成的DNA子链的一个组成部分。因此所产生的新链便具有已发生突变的碱基序列。为了使目的基因的特定位点发生突变,所设计的寡核苷酸引物的序列除了所需的突变碱基外,其余的则与目的基因编码链的特定区段完全互补。
2)诱变过程
M13寡核苷酸诱变过程的主要步骤如下(图13-7)。①正链DNA的合成:目的基因克隆到M13噬菌体中,制备含有目的基因的M13单链DNA,即正链DNA。②突变引物的合成:用化学法合成带错配碱基的诱变剂寡核苷酸片段,即寡核苷酸引物,其中除了含有特殊的突变碱基外,其他碱基与目的DNA的适当区域互补。③异源双链DNA分子的制备:将突变引物DNA与含目的基因的M13单链DNA混合退火,使引物与待诱变核苷酸部位及其附近形成一小段具有碱基错配的异源双链DNA。在Klenow DNA聚合酶催化下,引物链便以M13单链DNA为模板继续延长,直至合成全长的互补链,然后由T4 DNA连接酶封闭缺口,最终在体外合成出闭环异源双链的M13 DNA分子。④闭环异源双链DNA分子的富集和转化:在体外合成异源双链的M13 DNA分子后,尚余有单链M13噬菌体DNA或具裂口的双链M13 DNA分子,转化大肠杆菌后,也会增殖而产生很高的转化本底。故转化前应使用S1核酸酶处理法或碱性蔗糖梯度离心法,降低本底,使闭环的异源双链的M13 DNA分子得到富集。然后将富集的闭环异源双链的M13 DNA分子转化给大肠杆菌细胞后,产生出同源双链DNA分子。⑤突变体的筛选:闭环异源双链DNA分子转化大肠杆菌后可产生野生型和突变型两种转化子,两者混合存在,故须进行筛选以获得突变型转化子。常用筛选方法有链终止序列分析法、限制位点法、杂交筛选法和生物学筛选法,其中杂交筛选法最简单,也最有用。在杂交实验中,以诱变剂寡核苷酸为探针,在不同温度下进行噬菌体斑杂交,选择突变体克隆,由于探针与野生型DNA之间存在着碱基错配,而与突变型则完全互补,于是可以根据两者杂交稳定性的差异,筛选出突变型的噬菌斑。⑥基因的鉴定:对突变体DNA进行序列分析,检测突变体的序列结构特点,有助于确定在诱变过程中是否引入其他偶然错配。
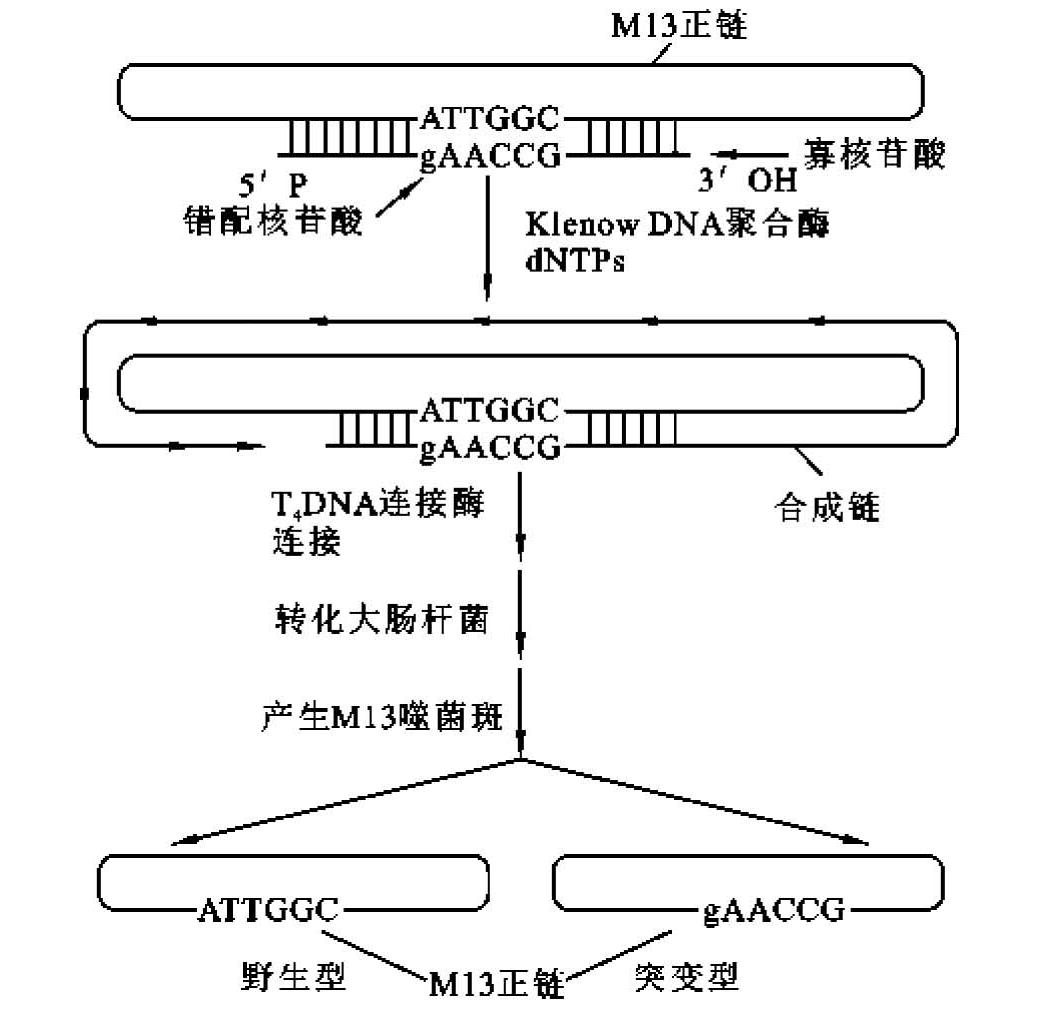
图13-7 寡核苷酸诱变过程示意图
(引自刘贤锡,2002)
3)Kunkel定点诱变法
体外DNA合成往往是不完全的,所以部分合成的DNA分子必须通过蔗糖密度梯度离心法除去。理论上来说,DNA是半保留复制的,应用寡核苷酸定点诱变时,所形成的噬菌体中携带突变基因的应为一半。但实际上由于宿主错配修复的反选择及其他技术上的原因,通常只有1%~5%的噬菌斑含有突变基因的噬菌体。因此,为了获得更多含有突变噬菌体的噬菌斑,必须提高突变体的比率。目前已有多种改良的寡核苷酸定点诱变方法,此处将简单介绍Kunkel在1985年建立的方法。
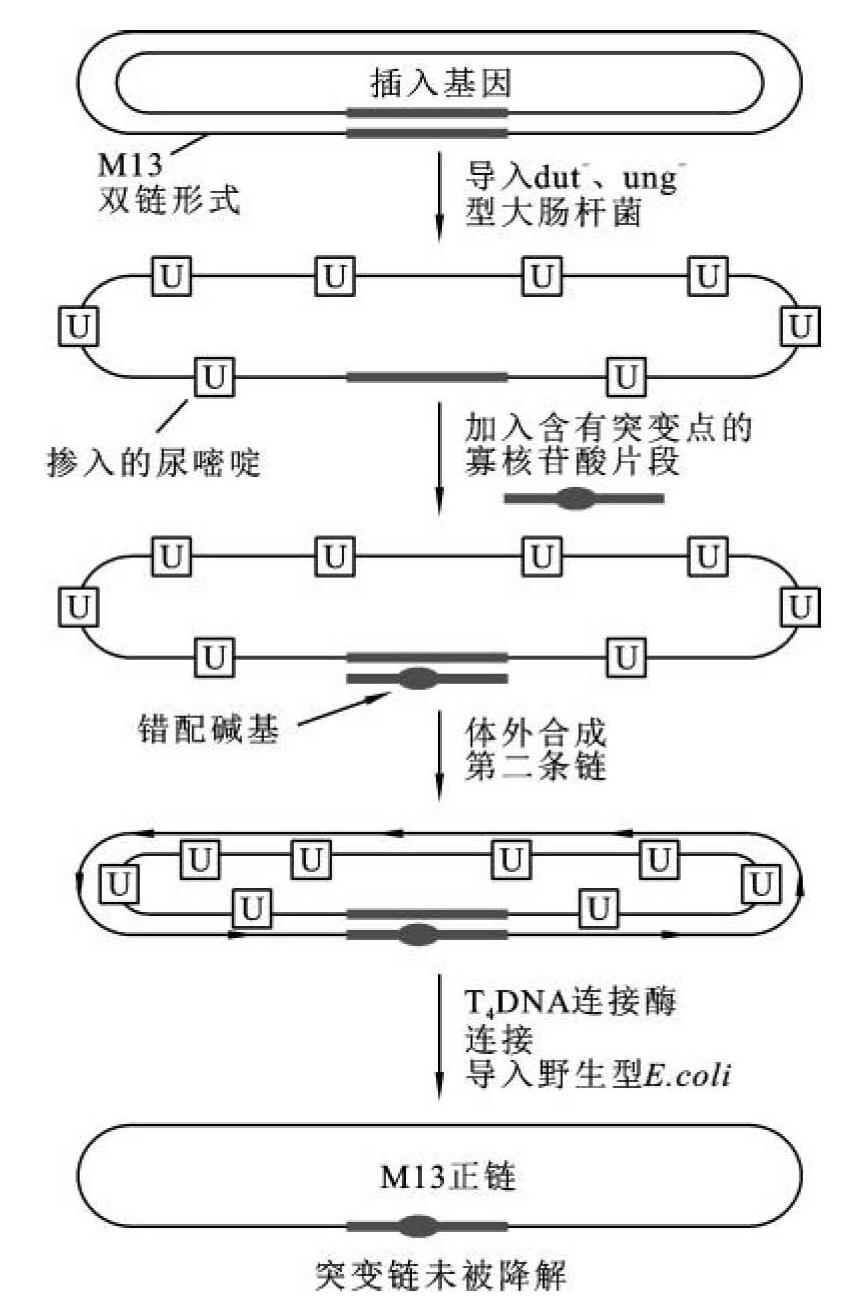
图13-8 Kunkel定点诱变示意图
(引自刘贤锡,2002)
Kunkel定点诱变法是一种通过筛除含有尿嘧啶的DNA模板链进行的寡核苷酸定点诱变法。它的基本原理是,将待突变的基因克隆入RF-M13 DNA载体上,导入具有dUTP酶(dut)和N-尿嘧啶脱糖苷酶(ung)双缺陷的大肠杆菌(dut-、ung-)菌株中。dut缺陷导致细胞内dUTP水平上升,并在DNA复制时,部分取代dTTP进入DNA新生链中。又由于ung缺陷,掺入DNA的dUTP残基不能除去。由这种大肠杆菌菌株产生的M13单链DNA大约有1%的T被U取代,以此为模板链,利用含有突变位点的寡核苷酸片段引导合成互补链。双链DNA导入正常的大肠杆菌中,其尿嘧啶N-糖基化酶除去DNA链上的尿嘧啶碱基。结果原来的M13模板链被降解。只有突变链因不含U,被保留下来(图13-8),这种方法产生的M13噬菌体中含有突变DNA的比率大大增加。
2.PCR定点诱变
寡核苷酸诱变方法所使用的M13噬菌体系统,操作起来比较烦琐,诱变周期较长。随着PCR技术的不断发展,人们将其应用在诱变技术中,使得此技术得以简化。
PCR定点诱变又称为PCR寡核苷酸定点诱变,该法具有简单、快捷的特点,其基本操作程序(图13-9)如下。①将待诱变靶基因克隆到质粒载体上,并分装到两支反应管中。②在每一支反应管中加入两种特定的引物,其中引物1和3均含有错配核苷酸,但两种引物分别与质粒DNA的不同链不完全互补,引物2和4均不含错配核苷酸,两者分别与质粒DNA的引物1和2杂交链的互补链完全互补。③进行PCR扩增获得含有突变碱基的线形质粒DNA。④将两支反应管中的线形质粒DNA混合,再经过变性和复性,一支反应管中的一条链和另一支反应管中的互补链杂交,通过两个黏性末端形成带有缺口的环状DNA分子。⑤转化大肠杆菌,环状DNA分子的缺口可被大肠杆菌修复。如果同一支反应管中的两个互补链又互相杂交,则继续形成线状DNA分子,在大肠杆菌中不稳定,易被降解。该方法把特异突变点导入克隆基因,无须把基因插入M13中,即可在大肠杆菌中进行表达。(https://www.daowen.com)
3.盒式诱变
盒式诱变(cassette mutagenesis)是一种定点诱变技术,其方法是利用一段人工合成的具有突变序列的双链寡核苷酸片段,取代野生型基因中相应序列(图13-10),这种诱变的双链寡核苷酸片段是由两条人工合成的寡核苷酸链组成的,当它们退火时,会按照设计要求产生出克隆需要的黏性末端。这些合成的寡核苷酸片段就好像不同的盒式录音磁带,可随时插入已制备好的载体分子(“录音机”)上,便可以获得数量众多的突变体,故称为盒式诱变。该方法的优点是简单易行,突变效率高。
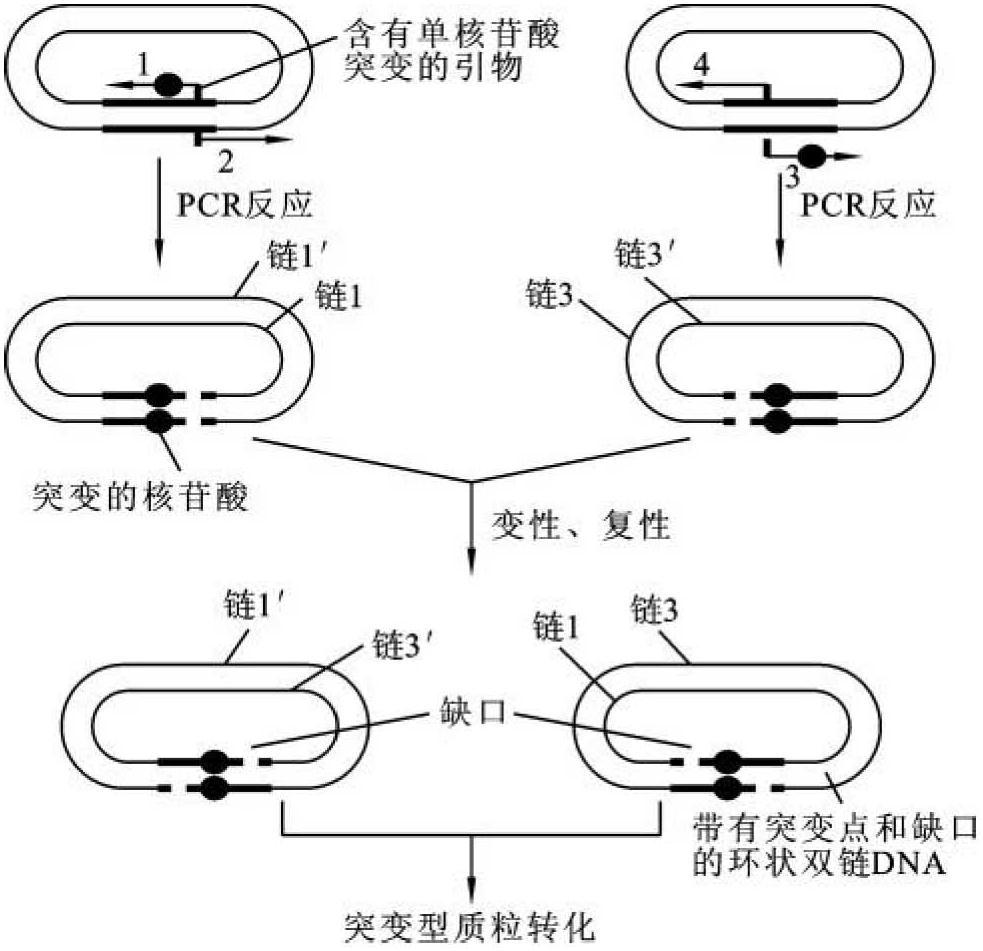
图13-9 PCR进行寡核苷酸定点诱变的示意图
(引自刘贤锡,2002)
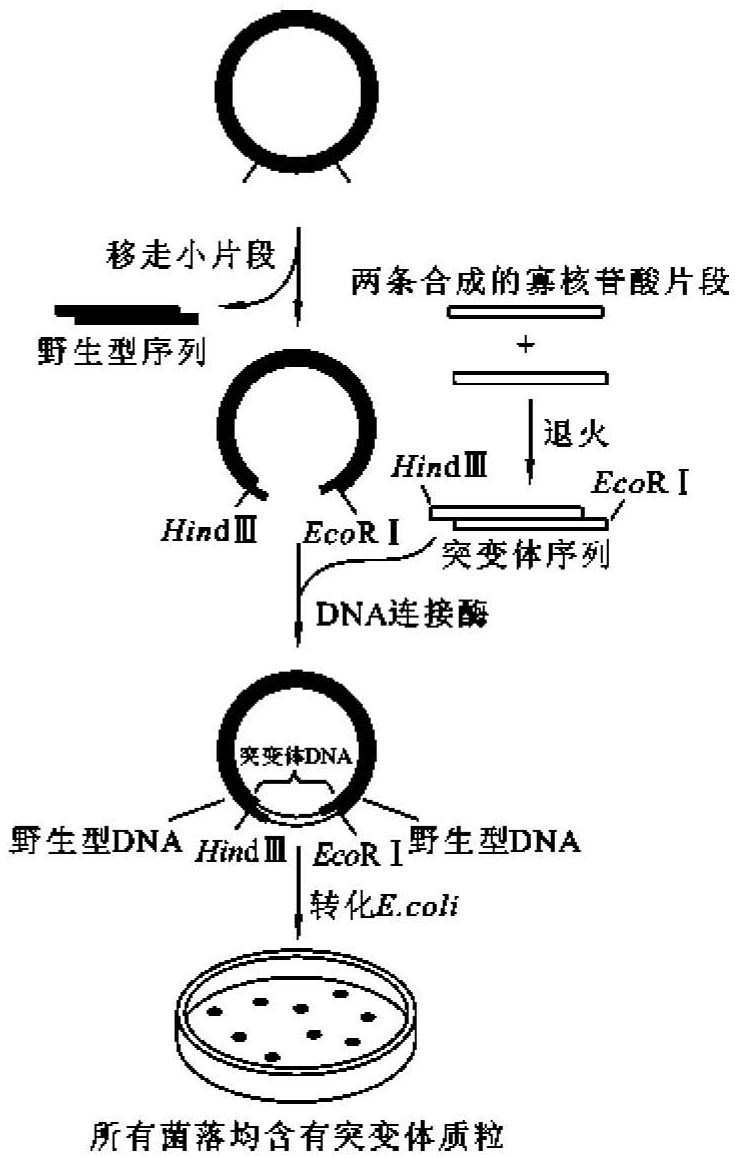
图13-10 盒式诱变示意图
(引自刘贤锡,2002)
4.随机诱变
定位诱变通常需要了解目的序列的详细情况,当缺乏这方面的资料和信息时,定位诱变方法的利用就受到限制,在这种情况下,利用随机诱变方法,在目的序列中产生突变,仍可以用于研究目的蛋白或目的核酸序列的结构和功能。
随机诱变(random mutagenesis)的工作原理(图13-11)是,将待突变基因克隆到一个载体的特定位点上,其下游紧接着是两个限制性核酸内切酶的酶切位点(RE1、RE2),前一酶切位点是5'突出(3'凹陷)的单链末端,后一酶切位点是3'突出(5'凹陷)的单链末端。然后用大肠杆菌核酸外酶(exonuclease Ⅲ,Exo Ⅲ)处理酶切缺口。ExoⅢ的主要活性是催化双链DNA自3'-羟基端逐一释放5'-单核苷酸,其底物是线状双链DNA或有缺口的环状DNA,而不能降解单链或双链DNA的3'突出末端。因此,当用ExoⅢ处理时,则可逐一水解3'凹陷末端。在适当时机,终止ExoⅢ的酶切反应,缺口用Klenow DNA聚合酶补平,底物为四种dNTP,再加上一种脱氧核苷酸的类似物。在缺口填补过程中,这个类似物会掺入DNA链上的一处或多处。再用S1核酸酶处理单链末端,形成平头末端,并用T4DNA连接酶连接。这种重组质粒转化大肠杆菌后,50%的基因上携带错配的碱基,导致位点变异。随机诱变的缺点是必须检测每个克隆,看哪一个产生了具有期望特性的蛋白质。这种检测不是一个简单的工作,但这是发现有新特性蛋白质的唯一方法。一旦发现了有潜在优点的突变体,通过确定哪个位点突变了可以确定克隆基因的序列。
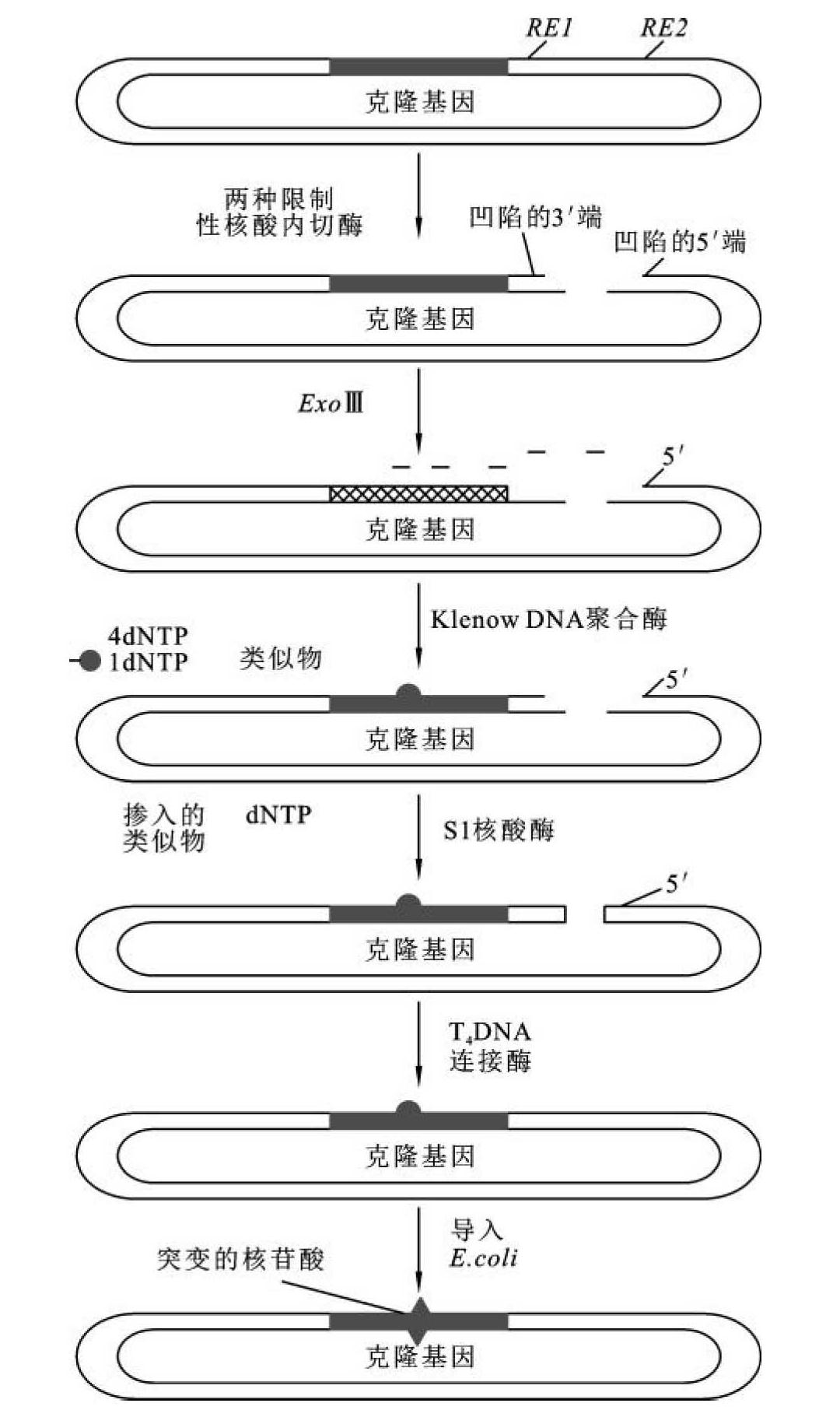
图13-11 随机诱变示意图
(引自刘贤锡,2002)