参考文献
第13章 蛋白质工程
【本章简介】 蛋白质工程是在重组DNA技术应用于蛋白质结构与功能研究之后发展起来的一门新兴学科。所谓蛋白质工程,就是通过对蛋白质已知结构和功能的了解,借助计算机辅助设计,利用基因定点诱变等技术,特异性地对蛋白质结构基因进行改造,产生具有新特性的蛋白质的技术。蛋白质工程是在遗传工程取得的成就的基础上,融合蛋白质结晶学、蛋白质动力学、计算机辅助设计和蛋白质化学等学科而迅速发展起来的一个新兴研究领域,它开创了按照人类意愿设计制造符合人类需要的蛋白质的新时期,因此,被誉为“第二代遗传工程”。蛋白质工程的出现,为认识和改造蛋白质分子提供了强有力的手段。
牛胰岛素是一种蛋白质分子,它的化学结构于1955年由英国科学家Sanger测定、阐明,即牛胰岛素分子是一条由21个氨基酸组成的A链和另一条由30个氨基酸组成的B链,通过两对二硫链联结而成的一个双链分子,而且A链本身还有一对二硫键。1965年我国率先人工合成结晶牛胰岛素,且其结晶形状和酶切图谱与天然物相同。人工合成结晶牛胰岛素是科学上的一次重大飞跃,它标志着人工合成蛋白质时代的开始,是生命科学发展史上一个新的重要里程碑。1978年,美国Hutchison使用了Lederberg于1960年推荐使用的寡脱氧核糖核苷酸作为体外诱变剂,成功地实现了定位突变(site-directed mutagenesis)试验,培育出多种具有生物学特性的突变株。1981年,美国Gene公司厄尔默(K.Ulmer)则将此定位突变试验冠以“蛋白质工程”(protein engineering)。经过多年的发展,蛋白质工程已经成为生命科学中的一个重要分支,它通常指通过蛋白质化学、蛋白质晶体学和蛋白质动力学的研究获取关于蛋白质物理、化学等方面的信息,在此基础上对编码该蛋白质的基因进行有目的的设计改造,并通过基因工程等手段将其进行表达和分离纯化,最终将其投入实际应用。蛋白质工程在食品、医药、工业等方面取得巨大成果。比如,抗体不仅在哺乳动物机体中担负着重要的体液免疫功能,还在医学、生物学免疫诊断中被广泛地应用。已证明了抗体是一类免疫球蛋白,并相继阐明了抗体产生及其多样性的细胞和分子机制,使免疫学研究成为生命科学前沿领域。同时抗体的制备技术也经历着一次又一次革命,由血清抗体到杂交瘤单克隆抗体,再到基因工程抗体库技术,可谓日新月异。
蛋白质工程研究在过去的30余年间发展迅速,已取得一批较好成果,并开始应用于医学、农业、轻工等各个领域,产生了较大的经济和社会效益。2000年,人类基因组的工作草图宣告完成,标志着人类跨进21世纪历史新纪元之际,生命科学也迎来了一个崭新的时代,即后基因组时代(post-genome era)。在后基因组时代,生物学的中心任务是揭示基因组及其所包含的全部基因的功能,并在此基础上阐明生命体的遗传、进化、发育、生长、衰老和死亡的基本生物学规律,以及与人类健康和疾病相关的生物学问题。由于基因的功能最终总是通过其表达产物蛋白质来实现的,因此,在人类基因组测序之后进一步集中研究蛋白质的结构及功能,是揭示基因组功能,阐释生命体重要生命活动规律和生命现象本质的基本途径,也是阐释疾病发生与发展的分子机理进而战胜疾病的重要途径。而研究蛋白质的结构和功能,并在此项研究基础上人工改造蛋白质的结构,获得我们所需要的活性蛋白质,正是蛋白质工程的主要任务和目标。
13.1 蛋白质工程的理论基础、诞生和发展
13.1.1 蛋白质工程的理论基础
酶的专一性强,在温和条件下有效地催化化学反应的能力使酶的应用日益广泛。药品、化学、食品工业及分析服务行业是酶开发的重要领域。目前已知的酶有8000多种,至少有2500种有可能应用,而目前国际上工业用酶约为50种,以吨级出售的仅20余种,可见,酶的开发利用尚有较大潜力。妨碍酶开发利用的主要原因如下:①生物材料中酶含量甚少,用传统酶蛋白的分离纯化成本太高;②酶蛋白分子结构的稳定性差,酸性或碱性过强、高温、氧化等因素均可破坏其结构,使其丧失生物活性,因而在工业加工条件下,酶的半衰期短,利用率较低;③酶催化活性的最适pH值及底物专一性的范围较窄,与工业应用的要求有较大差距。因此,要想扩大酶蛋白的开发利用,需要建立适当的方法,以改善酶蛋白的生物特性,使之适合于工业应用的要求。
随着基因工程理论和技术的发展,已经能克隆特异酶基因,并令其在适宜宿主菌中表达,使酶的产量大大提高,因而降低酶纯化成本的问题已基本解决。重要的是提高酶蛋白的稳定性,改良其生物学特性。
稳定蛋白质空间构象的主要因素是蛋白质分子众多基团间的相互作用,如肽键和侧链间的氢键、带有不同电荷的侧链间的静电作用、极性氨基酸残基侧链间的偶极作用、疏水残基间的疏水作用以及分子内的二硫键等。蛋白质结构与功能研究表明,上述稳定蛋白质空间构象的因素是由蛋白质一级结构中某一个或某一段氨基酸序列决定的,人工改变或修饰这些氨基酸残基,有可能增加蛋白质的稳定性,又不影响其生物学活性。如野生型枯草芽孢杆菌蛋白酶的218位Asn是与酶活性有关的氨基酸残基,若将其变成Ser,用65℃的失活半衰期衡量,从59 min增加到223 min,酶的稳定性显著增加。再如氧化失活使枯草芽孢杆菌蛋白酶在工业上的应用受到严重限制。很早就有人证实枯草芽孢杆菌蛋白酶在少量H2O2作用下很快失活是由于埋藏在催化部位Ser221邻近的Met222氧化成硫氢化物。1985年Wells证明若用其他氨基酸取代Met222,可以提高酶的氧化稳定性而又保持其高催化活性。
以上以酶为例介绍了蛋白质工程的重要性。实际上,在多肽药物、抗体以及其他活性蛋白质的基础与应用研究中,同样存在着蛋白质结构的改造问题。如白细胞介素-2(IL-2)是一种免疫反应调节因子,在医学上具有广泛的用途。结构研究证明,IL-2是由133个氨基酸残基组成的多肽,有3个半胱氨酸残基、1个二硫键,而125位上的半胱氨酸残基处于游离状态,这可能与其结构的稳定性有关。在分离纯化IL-2的过程中,发现在产品纯化和恢复活性过程中,IL-2多肽链中的3个半胱氨酸残基之间易发生二硫键错配,致使整个多肽活性降低。对此,科学家通过定点诱变将编码链上编码125位半胱氨酸残基的密码子TGT转换成TCT或GCA,也就是把半胱氨酸残基转换成丝氨酸残基或丙氨酸残基,结果可以避免二硫键的错配,使产品IL-2的活性提高了7倍。可见,改造蛋白质的空间结构以改变蛋白质的某些生物学特性,在拓宽蛋白质的用途、推动生物技术产业化方面至关重要。而蛋白质精细结构的改造,只有利用蛋白质工程技术才能得以实现。
蛋白质工程的重要目标之一,是通过改造蛋白质的结构来提高其开发利用的价值。这就是说,蛋白质的结构改变了,其生物学功能不能变。蛋白质的功能是由其结构决定的,蛋白质的结构改变了,其生物学功能能保持不变吗?蛋白质结构与功能关系的研究表明,蛋白质功能部位的几个氨基酸残基决定着蛋白质的功能,但是这几个负责功能的氨基酸残基必须处于一个极其精密的空间状态下才能发挥功能。这就是说,只要能维持蛋白质结构域的必需氨基酸残基及空间构象不变,其功能域的生物学活性就会保持不变。变异研究已经证明,许多蛋白质分子中多个氨基酸残基被改变,但其生物学功能不受影响或只受极少的影响。事实上,在许多个体中已发现一些变异蛋白质,伴有氨基酸残基的插入、丢失或替换,但蛋白质仍保持正常功能。如前所述,将枯草芽孢杆菌蛋白酶催化部位Ser221邻近的Met222置换,可以提高该蛋白酶的抗氧化性,又能保持催化活性,就是一个典型的例子。随着蛋白质三维结构分析技术的不断发展,借助于计算机对蛋白质结构域与功能域信息的处理及设计,采用定点诱变技术从基因水平改变氨基酸残基编码顺序,即可获得与天然蛋白质的理化性质相异而生物活性相似的突变蛋白质。
生物技术的兴起使得分子生物学的理论与工程实践紧密结合。20世纪70年代初期,重组DNA技术诞生,并成功地应用于基因操作,从而产生了基因工程。而基因工程的诞生,特别是重组DNA技术和基因定点诱变等技术的建立,使我们有可能从基因水平改造蛋白质分子中氨基酸序列,为蛋白质工程的诞生奠定了技术基础。所谓基因定点诱变,就是在DNA水平上,通过对蛋白质结构基因中某个或某些氨基酸残基编码序列加以改变,从而改变蛋白质结构。该技术不仅可用于改造天然蛋白质,而且可以用于确定蛋白质中每一个氨基酸残基在结构和功能上的作用,以收集有关氨基酸残基线形顺序与其空间构象和生物学活性之间的对应关系,为人工改造蛋白质提供理论依据。
总之,蛋白质结构和动力学研究是蛋白质结构与功能关系研究的重要手段,而改造蛋白质结构,则依靠基因工程,基因工程的发展从技术上提供了改变蛋白质个别氨基酸残基或肽段的手段,使结构改变已能在实验室实施,两者结合则产生了蛋白质工程。
13.1.2 蛋白质结构测定与结构预测
一种生物体的基因组规定了所有构成该生物体的蛋白质,基因规定了组成蛋白质的氨基酸序列。虽然蛋白质由氨基酸残基的线状序列组成,但是,它们只有折叠成特定的空间构象才能具有相应的活性和相应的生物学功能。了解蛋白质的空间结构不仅有利于认识蛋白质的功能,也有利于认识蛋白质是如何执行其功能的。确定蛋白质的结构对于生物学研究是非常重要的。目前,蛋白质序列数据库的数据积累的速度非常快,但是已知结构的蛋白质相对比较少。
尽管蛋白质结构测定技术有了较为显著的进展,但是通过实验方法确定蛋白质结构的过程仍然非常复杂,代价较高。因此,实验测定的蛋白质结构比已知的蛋白质序列要少得多。另一方面,随着DNA测序技术的发展,人类基因组及更多的模式生物基因组已经或将要被完全测序,DNA序列数量将会剧增,而由于DNA序列分析技术和基因识别方法的进步,可以从DNA推导出大量的蛋白质序列。这意味着已知序列的蛋白质数量和已测定结构的蛋白质数量(如蛋白质结构数据库PDB中的数据、布鲁克海文蛋白质数据库关于生物大分子三维结构的数据)的差距将会越来越大。人们希望产生蛋白质结构的速度能够跟上产生蛋白质序列的速度,或者减小两者的差距。那么如何缩小这种差距呢?不能完全依赖现有的结构测定技术,需要发展理论分析方法,这对蛋白质结构预测提出了极大的挑战。20世纪60年代后期,Anfinsen首先发现去折叠蛋白质(或者说变性蛋白质)在允许重新折叠的实验条件下可以重新折叠到原来的结构,这种天然结构对于蛋白质行使生物功能具有重要作用,大多数蛋白质只有在折叠成其天然结构的时候才具有完全的生物活性。自从Anfinsen提出蛋白质折叠的信息隐含在蛋白质的一级结构中,科学家们对蛋白质结构的预测进行了大量的研究,分子生物学家将有可能直接运用适当的算法,从氨基酸序列出发,预测蛋白质的结构。
蛋白质结构预测的目的是利用已知的一级序列来构建出蛋白质的立体结构模型,从而在此基础上进行结构与功能的研究以及蛋白质分子设计工作。一般在给定蛋白质一级序列后,首先要做的是进行序列对比,在蛋白质序列数据库或晶体结构数据库中寻找与之同源的蛋白质。如果找出的同源蛋白质已经有了晶体结构,就可以利用同源蛋白质结构预测的方法构建出该蛋白质的结构模型。如果没有已知的同源蛋白质结构,经典的做法是先进行二级结构预测、超二级结构预测,再进行三级结构预测。由于二级结构预测准确度的限制,这种方法在多数情况下(已知很少的实验信息)难以完全成功,只能给出一些有用的参考信息。
1.序列同源性分析
蛋白质由22种氨基酸残基组成,假设一个蛋白质序列有350个残基,那么其可能的序列排列数应该是20350,这是一个巨大的数字,而实际上蛋白质序列的数目还远不止这个数字。但是,随着氨基酸序列的继续增加,人们越来越清楚地认识到,存在于地球生物体系中的蛋白质结构类型是有限的。实际上,小数目的基因对应的蛋白质结构,通过通常的方式“复制和修饰”得到扩展。基因复制的程度是不同的,从非常小的片段到超基因以至整个染色体。复制后的修饰主要采取核苷酸残基替代,从而导致氨基酸变化。因此,从理论上讲,一组由复制、突变而来的蛋白质序列之间或多或少地存在着同源性。也就是说,对于一个新序列,首先要做的就是对其进行序列分析,看其是否与某些已知的序列存在同源性,进而对其结构和功能有一个大概了解。如在蛋白质序列数据库PIR中,FASTA是整库搜寻程序,它能够在一个蛋白质或核酸序列库中找到与某个蛋白质或核酸同源的序列,也能够将某个蛋白质与整个核酸序列库进行同源性比较。其原理为:给定一个目标序列(核酸或蛋白质),然后将该序列与序列库中的所有序列进行比较,如果目标序列与数据库序列的同源得分较高,则认为两个序列是同源的,程序给出数据库序列的代码。分析这些同源序列的结构与功能,将有助于目标序列的研究。
此外,同源性分析还应用于以下几个方面:
(1)进行保守位点和活性位点的分析。对一组序列进行同源性分析,在各个序列中都保守的位点就是可能的活性位点,进而设计实验,确定其中的活性位点。
(2)在序列分析的基础上建立蛋白质之间的进化关系。
(3)在序列比较的基础上进行二级结构预测,可提高预测的准确度。二级结构预测的准确度一般不超过65%,因此,在对一个序列进行二级结构预测前,先挑选一组同源物,进行序列比较,然后列出各个序列的二级结构(已知的或预测的),以此为参照,可提高二级结构预测的准确度。
(4)在序列分析的基础上可以预测同源蛋白质的三级结构。
(5)预测分析蛋白质的折叠模式。Bowie等分析蛋白质晶体结构数据库中已知结构蛋白质各残基的疏水度和表面积,以及二级结构与氨基酸残基的关系,得到一个氨基酸残基一级序列与三级序列的关系表。以此为基础进行序列比较,预测蛋白质的折叠模式。
由此可见,蛋白质序列的同源性分析是蛋白质结构预测和分子设计的基础。
2.双重序列对比
双重序列对比是两个序列的比较。两个功能类似的蛋白质,通过序列对比,可以确定其是否存在同源性;反之,两个具有同源性的序列,在功能上可能是类似的。在计算机辅助序列对比的方法中,比较经典的是Needle-Wunsch动力学比较方法。
1)原理
设有两个序列A、B,在其序列对比中,最小的比较单位是氨基酸残基,一个来自序列A,另一个来自序列B,序列对比就是寻找两个序列的最大匹配路径。最大匹配数定义为在允许两个序列有插入和删除的残基的基础上,序列A与序列B对上的最多残基数,这个数目可以通过由序列A、B组成的二维矩阵得到,如图13-1所示。

图13-1 Needle-Wunsch动力学比较方法原理示意图
(引自黄迎春,2009)
在图13-1中,两个序列交成一个二维矩阵,对每两个残基进行比较,可以从序列的一端开始,例如从末端(图的右下角)开始。假设求RR矩阵元数值,首先假设RR在打分矩阵中的得分为1,再求出这个矩阵元的下一行下一列的最大得分,两者加起来,即为该矩阵的最大得分。当最大得分不与该矩阵元相邻时,还应加上一个gap值,得到每个矩阵元的数值后,从序列的另一端开始沿最大矩阵元得分的途径将两个序列残基一一对应起来。
2)打分矩阵的gap值
自从1968年Dayhoff等报道了第一个PAM矩阵后,又出现了一系列的打分矩阵。在这些矩阵中,有的是根据氨基酸所对应的核苷酸的变异而得到的,如GC矩阵、GCM矩阵、GDM矩阵等,矩阵中的值一般是据此确定的:如果2个氨基酸所对应的3个核苷酸中有2个是相同的,则其相似值为2。有的矩阵是根据氨基酸的物理化学性质得到的,如Rao矩阵综合了5个物理参数,包括Chou-Fasman的α-螺旋、β-折叠和转角的倾向性参数;HYDOR矩阵考虑了氨基酸残基侧链的疏水性质。有的矩阵是根据氨基酸在一组相关蛋白质中相互间的替代关系得到的,如PAM矩阵、MD矩阵等。
在蛋白质三级结构的预测方法中,由同源物结构构建未知蛋白质结构是一种比较成熟的方法。在这种方法中,首先需要对同源蛋白质进行序列比较。但由于序列比较与结构叠合的结果往往有一些差异性,因此人们试图建立一些基于结构信息基础上的打分矩阵,称之为结构打分矩阵。例如,RIS打分矩阵是根据一组叠合的蛋白质结构中拓扑结构相应区域的氨基酸残基的取代关系得到的。SCM矩阵是根据氨基酸残基的主链二面角分布得到的:先计算所有已知结构的蛋白质中每个氨基酸残基的二面角分布(图13-1),比较22种残基的相似形,以此建立起不同残基间的结构替代关系。SCFm矩阵、SCFs矩阵是根据氨基酸残基的空间倾向性因子得到的:先统计所有已知结构的蛋白质中22种残基的主链及侧链空间倾向性因子,然后计算其相关系数,作为不同残基间结构相似性的量度指标。
在这些结构打分矩阵中,每个氨基酸残基代表一个结构单元,这样序列对比的结果可以反映蛋白质结构的相似性。但是,由于这些打分矩阵都来自统计结果,反映了蛋白质或其一个家族的共性,因此,将实际上在折叠过程中处于不同环境中的同种氨基酸残基看作同一种结构类型,在打分矩阵中得分最高。显然在这种结构比较中同时引入序列信息,序列对比的结果仅仅可以在一定程度上反映蛋白质结构的相似性,因此,称利用结构打分矩阵的序列比较为类结构比较。
在双重序列对比中,另一个需要的参数是gap值。gap对应于序列中的插入或删除。在gap处,序列对比的累计得分应扣除某个值。如果扣除某个值后,序列对比的累计得分仍是多条路径中得分最高的,那么在该处gap是允许的。在序列N末端或C末端的gap值为零,一个gap只能与一段序列相比,而不能与另一个gap相比。
3)序列对比的显著性得分
一对序列对比的最大得分是序列的氨基酸残基组成和两个序列之间关系的函数。怎样判断一对序列对比的最大得分是显著的,即两个序列间存在着可观的同源性?设立可能性实验,将两个序列分别重排。保持各自的氨基酸残基组成不变,然后进行序列对比;接下去重复进行重排一序列对比这一过程。如果次数足够多,就可以求出这一氨基酸残基组成的所有可能的序列对比的最大得分;如果两个序列的最大得分显著不同于重排序列的得分,就可以认为这两个序列间存在着某种程度的同源性。利用显著性得分这个概念来表示两个序列间的关系:
式中,A表示显著性得分;Sreal表示两个实际序列得分比较的最大得分;Srand表示将两个序列中的残基任意重排后进行比较的平均得分;D表示标准偏差。
一般在计算机中,残基重排—序列对比的过程重复100次就可以了。当两个序列的相同残基数比例高于20%时,实际序列对比的结果一般是可靠的。
4)双重序列对比实例
首先,给出打分矩阵,列于图13-2。利用这个打分矩阵,分别比较两对不同同源性的序列,如图13-3、图13-4所示。图13-3中的两个序列分别为人血红蛋白的β链和γ链,其序列同源性为73%。图13-4中的两个序列分别为人的肌红蛋白和腹足纲软体动物类的肌红蛋白,其序列同源性为22%。

图13-2 打分矩阵:依据氨基酸残基在一种相关蛋白质中相互间的替代关系(MD)
(引自黄迎春,2009)

图13-3 两个序列比较的结果一
(引自黄迎春,2009)

图13-4 两个序列比较的结果二
(引自黄迎春,2009)MYOH表示人肌红蛋白;MYCR表示腹足纲软体动物类的肌红蛋白;*表示相同残基,其余区域表示2个矩阵的序列对比结果相同。
3.多重序列对比
当研究一组蛋白质序列之间的关系时,双重序列对比往往引起一些不一致性。例如,当序列A与B比较时,在某些位置出现gap;当序列A与C比较时,gap的位置或许完全不同。为了克服这一缺点,又发展了一系列多重序列对比的方法。多重序列对比仍然以双重序列为基础。在进行多个序列比较之前,需先定出序列比较的顺序。一般来讲,序列同源性较高,序列比较的可靠性越大。因此在进行多重序列比较时,先根据序列同源性高低定出序列比较的顺序。
双重序列对比的显著性得分可以反映序列同源性的高低,但是,为了求显著性得分,需要重复多次序列重排,计算最大得分的过程。假设有6个序列,计算1对序列的显著性得分时将序列重排100次,那么,要分别求出每两个序列间的显著性得分,需进行![]() ×101=1515次序列对比,很费时间。Doolittle等证明,归一化的对比得分NAS值与显著性得分成正比,其中NAS定义为
×101=1515次序列对比,很费时间。Doolittle等证明,归一化的对比得分NAS值与显著性得分成正比,其中NAS定义为
NAS=两个序列的最大相似得分/最短的序列长度
这样可以根据NAS值来对序列排序,以进行多重序列对比。在双重序列对比的基础上,可得到一系列的对序列比较的NAS值。很显然,NAS值高的一对序列是同源性最好的序列,将它们作为多重序列对比中最先考虑的两个序列;然后挑选分别与这两个序列比较时NAS值较高的第三个序列;后面的序列选择以此类推,从而将整个序列按同源性高低排序。多重序列对比的方法有多种,这里介绍其中两种方法。
1)Feng-Doolittle方法
假设有一组排序后的序列为A,B,C,D,E,F,…,先对序列A、B进行比较。在对序列C进行比较时,分别计算序列对(A,C)和(B,C)的最大得分。如果序列(B,C)比较的得分高,则前三个序列比较的结果为ABC;同样再进行第四个序列D的比较,根据ABCD和CBAD的得分确定D的位置。以此类推。在序列比较的过程中,前一次比较中出现的gap在比较中仍然存在。图13-5是一组血红蛋白的比较结果,图中代码所代表的蛋白质列于表13-1。

图13-5 利用Feng-Doolittle方法对12个血红蛋白的序列比较
(引自黄迎春,2009)
表13-1 序列对比中用到的12个血红蛋白

(引自黄迎春,2009)
2)Barton-Sternberg方法
假设一组排序后的蛋白质序列为A,B,C,D,…,先比较同源性高的A、B两个序列。在对序列C进行比较时,须同时考虑A、B两个序列的残基。假设i是序列A1,A2,…,Ak-1等已经比较过的序列的一个残基位置,1≤i≤Lj(k-1),L为序列长度,j是序列Ak的一个残基位置,则相应矩阵元的得分为
以此为基础,将所有的序列对比起来,然后以对比好的序列为模板,比较所有这些蛋白质序列,如果残基的位置有变动,需再迭代一次,直至最后所有序列的所有残基位置不再变化。图13-6是一组血红蛋白的序列比较结果,图中代码所代表的蛋白质列于表13-1。

图13-6 利用Barton-Sternberg方法对12个血红蛋白的序列比较
(引自黄迎春,2009)
4.蛋白质二级结构的预测
蛋白质二级结构的预测不但是研究蛋白质折叠问题的主要内容之一,而且是获得新氨基酸序列结构信息的一般方法。目前预测蛋白质二级结构的方法有几十种,可以归纳为统计方法、基于已有知识的预测方法和混合方法。这些方法都假定蛋白质的二级结构主要是由邻近残基间的相互作用所决定的,然后通过对已知空间结构的蛋白质分子进行分析、归纳,制定出一套预测规则,并根据这些规则对其他已知或未知结构的蛋白质分子的二级结构进行预测。这里仅介绍统计方法中的Chou-Fasman方法。
Chou和Fasman在20世纪70年代,基于单个氨基酸残基统计的经验参数方法,通过统计分析,获得每个残基出现于特定二级结构构象的倾向性因子,进而利用这些倾向性因子预测蛋白质的二级结构。
每种氨基酸残基出现在各种二级结构中的倾向或者频率是不同的,例如Glu主要出现在α-螺旋中,Asp和Gly主要分布在转角中,Pro也常出现在转角中,但是绝不会出现在α-螺旋中。因此,可以根据每种氨基酸残基形成二级结构的倾向性或者统计规律进行二级结构预测。另外,不同的多肽片段有形成不同二级结构的倾向,例如:肽链Ala(A)-Glu(E)-Leu(L)-Met(M)倾向于形成α-螺旋,而肽链Pro(P)-Gly(G)-Tyr(Y)-Ser(S)则不会形成α-螺旋。
通过对大量已知结构的蛋白质进行统计,为每个氨基酸残基确定其二级结构倾向性因子。在Chou-Fasman方法中,这几个因子是Pα、Pβ和Pt,它们分别表示相应的残基形成α-螺旋、β-折叠和转角的倾向性。另外,每个氨基酸残基同时也有四个转角参数,即f(i)、f(i+1)、f(i+2)和f(i+3)。这四个参数分别对应于每种残基出现在转角第一、第二、第三和第四位的概率,例如,脯氨酸约有30%出现在转角的第二位,然而出现在第三位的概率不足4%。根据Pα和Pβ的大小,可将22种氨基酸残基分类,如谷氨酸、丙氨酸是最强的螺旋形成残基,而缬氨酸、异亮氨酸则是最强的折叠形成残基。除各个参数之外,还有一些其他的统计经验,如脯氨酸和甘氨酸最倾向于中断螺旋,而谷氨酸则通常倾向于中断折叠。
在统计得出氨基酸残基倾向性因子的基础上,Chou和Fasman提出了二级结构的经验规则,其基本思路是在序列中寻找规则二级结构的成核位点和终止位点。在具体预测二级结构的过程中,首先扫描待预测的氨基酸序列,利用一组规则发现可能成为特定二级结构成核区域的短序列片段,然后对成核区域进行扩展,不断扩大成核区域,直到二级结构类型可能发生变化为止,最后得到的就是一段具有特定二级结构的连续区域,下面是4个简要的规则。
(1)α-螺旋规则:沿着蛋白质序列寻找α-螺旋核,相邻的6个残基中如果有至少4个残基倾向于形成α-螺旋,即有4个残基对应的Pα>100,则认为是螺旋核。然后从螺旋核向两端延伸,直至四肽片段Pα平均值小于100为止。按上述方式找到的片段长度大于5,并且Pα平均值大于Pβ平均值,那么这个片段的二级结构就被预测为α-螺旋。此外,不容许Pro在螺旋内部出现,但可出现在C末端以及N末端的前三位,这也用于终止螺旋的延伸。
(2)-折叠规则:如果相邻6个残基中有4个倾向于形成β-折叠,即有4个残基对应的Pβ>100,则认为是折叠核。折叠核向两端延伸直至4个残基Pβ平均值小于100为止。若延伸后片段的Pβ平均值大于105,并且Pβ平均值大于Pα平均值,则该片段被预测为β-折叠。
(3)转角规则:转角的模型为四肽组合模型,要考虑每个位置上残基的组合概率,即特定残基在四肽模型中各个位置的概率。在计算过程中,对于从第i个残基开始的连续4个残基的片段,将上述概率相乘,根据计算结果判断是否是转角。如果f(i)×f(i+1)×f(i+2)×f(i+3)大于7.5×10.5,四肽片段Pt平均值大于100,并且Pt平均值同时大于Pα平均值以及Pβ平均值,则可以预测这样连续的4个残基形成转角。
(4)重叠规则:假如预测出的螺旋区域和折叠区域存在重叠,则按照重叠区域Pα平均值和Pβ平均值的相对大小进行预测,若Pα平均值大于Pβ平均值,则预测为螺旋;反之,预测为折叠。
Chou-Fasman方法原理简单明了,二级结构参数的物理意义明确,该方法中二级结构的成核、延伸和终止规则基本上反映了真实蛋白质中二级结构形成的过程。该方法的预测准确率在50%左右。
5.蛋白质三维结构的预测
1)三维结构的预测方法
生物信息学研究的一个主要目标是了解蛋白质序列与三维结构的关系,但是序列与结构之间的关系是非常复杂的。现已掌握了一些蛋白质序列与二级结构之间的关系,但是对于蛋白质序列与空间结构之间的关系了解得比较少。预测蛋白质的二级结构只是预测折叠蛋白的三维形状的第一步。一些结构不是很规则的环状区域,与蛋白质的二级结构单元共同堆砌成一个紧密的球状天然结构。生物化学研究中一个活跃领域就是了解引起蛋白质折叠的各种作用力。在蛋白质折叠过程中,一系列不同的力都起到重要作用,包括疏水作用、静电力、氢键和范德华力。疏水作用是影响蛋白质结构的重要因素。半胱氨酸之间共价键的形成在决定蛋白质构象中也起了决定性的作用。在一类称为伴侣蛋白的特殊蛋白质作用下,蛋白质折叠问题变得更复杂。伴侣蛋白通过一些未知的方式改变蛋白质的结构,但这些改变方式是很重要的,蛋白质三维结构的预测方法大致可以分为三类:①根据基本物理原理,用分子动力学(MD)模拟预测蛋白质的三维结构;②根据蛋白质同源性预测;③根据结构类型预测。
2)蛋白质分子动力学模拟预测
在蛋白质结构和功能研究中,经常使用分子动力学计算。分子动力学是一种计算机模拟技术,它的主要部分是求解与体系中每个原子相关的牛顿运动方程或薛定谔方程。蛋白质分子动力学是以蛋白质为研究体系的分子动力学。它用来模拟预测蛋白质结构的方法,现在主要是作为其他预测方法的补充手段和应用于结构优化。它的主要优点是可以利用有限的实验数据构造分子的结构模拟并研究它的能量与结构的动态变化,而这些数据对于用实验方法来确定结构是远远不够的。
3)同源模型化方法
根据蛋白质同源性进行预测是蛋白质三维结构预测的主要方法。对蛋白质数据库PDB分析可以得到这样的结论:任何一对蛋白质,如果两者的序列等同部分超过30%(序列比对长度大于80),则它们具有相似的三维结构,即两个蛋白质的基本折叠相同,只是在非螺旋和非折叠片层区域的一些细节部分有所不同。蛋白质的结构比蛋白质的序列更保守,如果两个蛋白质的氨基酸序列有50%相同,那么约有90%的α-碳原子的位置偏差不超过0.3 nm,这是同源模型化方法在结构预测方面成功的保证。同源模型化方法的主要思路如下:对于一个未知结构的蛋白质,首先通过序列同源分析找到一个已知结构的同源蛋白质,然后以该蛋白质的结构为模板,为未知结构的蛋白质建立结构模型。这里的前提是必须有一个已知结构的同源蛋白质。这个工作可以通过搜索蛋白质结构数据库来完成,如搜索PDB。同源模型化方法是目前一种比较成功的蛋白质三维结构预测方法。从上述方法介绍也可以看出,预测新结构是借助于已知结构的模板而进行的,选择不同的同源蛋白质,则可能得到不同的模板,因此最终得到的预测结果并不唯一。假设待预测三维结构的目的蛋白质为U(un-known),利用同源模型化方法建立结构模型的过程包括下述步骤:
(1)搜索结构模型的模板(T):同源模型化方法假设两个同源的蛋白质具有相同的骨架。为待预测的蛋白质建立模型时,首先按照同源蛋白质的结构建立模板T。所谓模板,是一个已知结构的蛋白质,该蛋白质与目的蛋白质(U)的序列非常相似。如果找不到这样的模板,则无法运用同源模型法。
(2)序列比对:将目的蛋白质(U)的序列与模板蛋白质(T)的序列进行比对,使U的氨基酸残基与T的残基匹配。比对中允许插入和删除操作。
(3)建立骨架:将模板结构的坐标拷贝到U,仅拷贝匹配残基的坐标。在一般情况下,通过这一步建立U的骨架。
(4)构建目的蛋白质的侧链:可以将模板相同残基的坐标直接作为U的残基坐标,但是对于不完全匹配的残基,其侧链构象是不同的,需要进一步预测。侧链坐标的预测通常采用已知结构的经验数据,如ROTAMER数据库的经验结构数据。ROTAMER数据库含有所有已知结构蛋白质中的侧链取向,按下述过程来使用ROTAMER数据库:从数据库中提取ROTAMER分布信息,取一定长度的氨基酸片段(对于螺旋和折叠取7个残基,其他取5个残基);在U的骨架上平移等长的片段,从ROTAMER数据库中找出那些中心氨基酸与平移片段中心相同的片段,并且两者的局部骨架尽可能相同,在此基础上从数据库中取局部结构数据。
(5)构建目的蛋白质的环区:在第(2)步的序列比对中,可能加入空位,这些区域常常对应于二级结构元素之间的环区,对于环区需要另外建立模型。一般也是采用经验方法,从已知结构的蛋白质中寻找一个最优的环区,复制其结构数据。如果找不到相应的环区,则需要用其他方法。
(6)优化模型:通过上述过程为U建立了一个初步的结构模型,在这个模型中可能存在一些不相容的空间坐标,因此需要进行改进和优化,如利用分子力学、分子动力学、模拟退火等方法进行结构优化。
当然,如果能够找到一系列与目的蛋白质相近的蛋白质的结构,得到更多的结构模板,则能够提高预测的准确性。通过多重序列比对,发现目标序列中与所有模板结构高度保守的区域,同时也能发现保守性不高的区域。将模板结构叠加起来,找到结构上保守的区域,为要建立的模型形成一个核心,然后按照上述方法构建目的蛋白质的结构模型。对于具有60%等同部分的序列,用上述方法建立的三维模型非常准确。若序列的等同部分超过60%,则预测结果将接近实验得到的测试结果。一般来说,如果序列的等同部分大于30%,则可以期望得到比较好的预测结果。
13.1.3 蛋白质工程的诞生与发展
蛋白质工程是20世纪80年代初诞生的一个新兴生物技术领域,其产生和发展涉及生物化学、生物物理学、分子生物学、分子遗传学、计算机科学和化学工程等学科,以及相关生物工程技术。由此可见,蛋白质工程的产生和发展是许多学科及相关生物工程技术相互融合、共同发展的结果,已成为生物工程技术的重要组成部分。
1.蛋白质工程与发酵工程
从广义上讲,发酵工程由上游工程、发酵工程(狭义)和下游工程三部分组成。上游工程是指优良菌株的选育及最适发酵条件的确立;发酵工程(狭义)是指在最适条件下,利用发酵罐进行细胞培养和生产代谢产物的技术;下游工程是指发酵产物的分离纯化。因此,发酵工程可为蛋白质工程提供优良稳定的上乘菌体,并可为蛋白质工程中前期材料的制备等奠定重要基础。
2.蛋白质工程与基因工程
蛋白质工程是从DNA水平改变基因入手,通过基因重组技术改造蛋白质或设计合成具有特定功能新蛋白质的新兴研究领域。也就是说,蛋白质的改造通常需要经过周密的分子设计,进而依赖基因工程获得突变型蛋白质。目前,基因工程已为实现蛋白质工程提供了基因克隆、表达、突变及活性检测等关键技术。
3.蛋白质工程与细胞工程
目前,细胞工程技术已为蛋白质工程提供了改良性状的微生物细胞以及稳定的动植物细胞系,以便更好地生产蛋白质;另外,细胞工程可通过细胞融合、转基因等技术改变生物的遗传性状或实现新型生物的构建,进而为蛋白质的生产提供更多良好的载体。
4.蛋白质工程与酶工程
酶工程是生物工程的重要组成部分,是酶的生产与应用的技术过程,是从应用的目的出发研究酶,利用酶的催化作用,在一定的生物反应器中,将相应的原料转化为所需要的产品。
酶工程的发展一般被认为是从第二次世界大战开始的。20世纪50年代开始,由微生物发酵液中分离出一些酶,制成酶制剂;60年代固定化酶及固定化细胞技术日益成熟;70年代后期以来,微生物学、遗传工程及细胞工程等相关技术被引入酶工程领域,促进了酶工程的发展。同时由于大部分酶的化学本质是蛋白质,因此酶工程与蛋白质工程的发展又有着密不可分的联系。
5.蛋白质工程与生物信息技术(https://www.daowen.com)
生物信息学是生物学与信息技术的交叉学科。该学科包含对核酸、蛋白质序列和蛋白质结构的信息处理,有助于对生物的基因组与蛋白质组的理解。生物信息学的概念是在1956年美国田纳西州盖特林堡召开的“生物学中的信息理论研讨会”上产生的,但直到20世纪80—90年代,随着生物科学技术的迅猛发展和数据资源的指数式增长以及计算机科学技术的进步,生物信息学才获得突破性进展。
生物信息学的研究内容主要包括生物信息的收集、存储、管理和提供,基因组序列信息的提取和分析,功能基因组相关信息的分析,生物大分子的结构模拟和药物设计等方面。利用生物信息学可以进行蛋白质结构的预测,其目的就是利用已知的一级序列来构建蛋白质的立体结构模型(包括二级和三级结构预测)。目前,已有大量的有关根据序列预测蛋白质二级结构的文献资料,大致可分为两类:根据单一序列预测二级结构;根据多序列预测二级结构。三级结构预测则需要利用数据库中已知结构的序列进行比对。
6.蛋白质工程与其他相关技术
结构分析和遗传物质的研究在蛋白质工程的发展中作出了重要的贡献。1912年Laue曾预言,晶体是X射线的天然衍射光栅;随后Bragg父子开创了X射线晶体学,并成功地测定了一些相当复杂的分子以及蛋白质的结构。20世纪中X射线衍射技术被正式应用到蛋白质的研究领域。1954年英国晶体学家Perute等提出,在蛋白质晶体中引入重原子的同晶置换法可用来测定蛋白质的晶体结构。1955年Sanger完成了胰岛素的氨基酸序列的测定,接着英国晶体学家Kendrew和Perute在X射线分析中应用重原子同晶置换技术和计算机技术,并分别于1957年和1959年阐明了鲸肌红蛋白和马血红蛋白的立体结构。1960年Kendrew等首次测出肌红蛋白的三维结构;1965年中国科学家合成了有生物活性的胰岛素,首先实现了蛋白质的人工合成。噬菌体感染宿主后半小时内即可复制出几百个同样的子代噬菌体颗粒,因此噬菌体是研究生物体自我复制的理想材料。到20世纪60年代中期,对于DNA自我复制和转录生成RNA的一般性质已基本清楚,基因的奥秘也随之开始解开了。进入20世纪70年代,由于重组DNA研究的突破,基因工程已开始在实际应用中“开花结果”,根据人的意愿改造蛋白质结构的蛋白质工程也已经成为现实。20世纪80年代美国Ulmer在《Science》期刊上发表了以“Protein Engineering”为题的专论,被视为蛋白质工程诞生的标志。
13.2 蛋白质工程的关键技术
对于存在于自然界的任何蛋白质来说,采用重组DNA技术克隆其基因,在特定宿主细胞中进行表达,可以获得运用于商业用途的纯化的产品。然而,这些蛋白质的理化特性常常不能适应工业用途。从生长于非常环境中的生物体中获得基因,可表达获得适用于特定目的的蛋白质。例如,从生存于90℃温泉中的Bacillus stearothermophilus分离获得α-淀粉酶基因,经表达可获得耐高温的α-淀粉酶,该酶可用于从淀粉制造乙醇的工业生产。传统诱变技术,也可以获得能编码具有期望特性的蛋白质的突变基因,并获得突变蛋白质。然而,在传统诱变方法中,经化学诱变剂或物理诱变剂处理的生物体,它的任何基因都有可能发生突变,而目的基因的突变频率又可能相当低,给突变体的筛选工作造成了很大的麻烦,而且所获得的突变蛋白质常常因其氨基酸的改变而导致蛋白质活性下降,因而传统诱变方法实用价值不大。
随着分子生物学技术的进步,建立起通过改变克隆基因中的特定碱基,从而改造蛋白质的技术,称为蛋白质工程。通过蛋白质工程,人们可以随心所欲地改造蛋白质的结构,从而改变蛋白质的理化性质和生物学功能。例如,可以利用蛋白质工程改变酶的Km(Michaelis常数)、vmax(酶催化反应的最大速度)值,酶促反应的最适温度、最适pH值,酶促反应的特异性以及酶蛋白的稳定性等。蛋白质工程与基因定点诱变有着不解之缘,故本节在重点介绍定点诱变原理和技术的基础上,举例介绍一下蛋白质工程的应用。
13.2.1 定点诱变
随着分子生物学技术的发展,特别是基因克隆技术的应用,分离并研究单基因的结构与功能已成为一种常规的工作。与此相适应,基因诱变技术也有了极大的发展。现在,不仅能够对多细胞或是有机体进行诱变处理,并从成千上万突变群体中筛选出期望的突变体,而且能在体外试管中通过碱基取代、插入或缺失使基因DNA序列中任何一个特定的碱基发生改变。这种体外特异性改变某个碱基的技术,叫做定点诱变(site-directed mutagenesis)。它具有简单易行、重复性高等优点,现已发展成为基因操作的一种技术。这种技术不仅适用于基因结构与功能的研究,还可通过改变基因的密码子来改造天然蛋白质。目前已发展的定点诱变方法主要有M13寡核苷酸诱变、PCR诱变、盒式诱变及随机诱变等,下面将逐一予以讨论。
1.M13寡核苷酸诱变
1)原理
寡核苷酸定点诱变技术所依据的原理是按照体外重组DNA技术,将待诱变的目的基因插入M13噬菌体上,制备此种含有目的基因的M13单链DNA,即正链DNA。再使用化学合成的含有突变碱基的寡核苷酸短片段作为引物,启动单链DNA分子进行复制,随后这段寡核苷酸引物便成为新合成的DNA子链的一个组成部分。因此所产生的新链便具有已发生突变的碱基序列。为了使目的基因的特定位点发生突变,所设计的寡核苷酸引物的序列除了所需的突变碱基外,其余的则与目的基因编码链的特定区段完全互补。
2)诱变过程
M13寡核苷酸诱变过程的主要步骤如下(图13-7)。①正链DNA的合成:目的基因克隆到M13噬菌体中,制备含有目的基因的M13单链DNA,即正链DNA。②突变引物的合成:用化学法合成带错配碱基的诱变剂寡核苷酸片段,即寡核苷酸引物,其中除了含有特殊的突变碱基外,其他碱基与目的DNA的适当区域互补。③异源双链DNA分子的制备:将突变引物DNA与含目的基因的M13单链DNA混合退火,使引物与待诱变核苷酸部位及其附近形成一小段具有碱基错配的异源双链DNA。在Klenow DNA聚合酶催化下,引物链便以M13单链DNA为模板继续延长,直至合成全长的互补链,然后由T4 DNA连接酶封闭缺口,最终在体外合成出闭环异源双链的M13 DNA分子。④闭环异源双链DNA分子的富集和转化:在体外合成异源双链的M13 DNA分子后,尚余有单链M13噬菌体DNA或具裂口的双链M13 DNA分子,转化大肠杆菌后,也会增殖而产生很高的转化本底。故转化前应使用S1核酸酶处理法或碱性蔗糖梯度离心法,降低本底,使闭环的异源双链的M13 DNA分子得到富集。然后将富集的闭环异源双链的M13 DNA分子转化给大肠杆菌细胞后,产生出同源双链DNA分子。⑤突变体的筛选:闭环异源双链DNA分子转化大肠杆菌后可产生野生型和突变型两种转化子,两者混合存在,故须进行筛选以获得突变型转化子。常用筛选方法有链终止序列分析法、限制位点法、杂交筛选法和生物学筛选法,其中杂交筛选法最简单,也最有用。在杂交实验中,以诱变剂寡核苷酸为探针,在不同温度下进行噬菌体斑杂交,选择突变体克隆,由于探针与野生型DNA之间存在着碱基错配,而与突变型则完全互补,于是可以根据两者杂交稳定性的差异,筛选出突变型的噬菌斑。⑥基因的鉴定:对突变体DNA进行序列分析,检测突变体的序列结构特点,有助于确定在诱变过程中是否引入其他偶然错配。
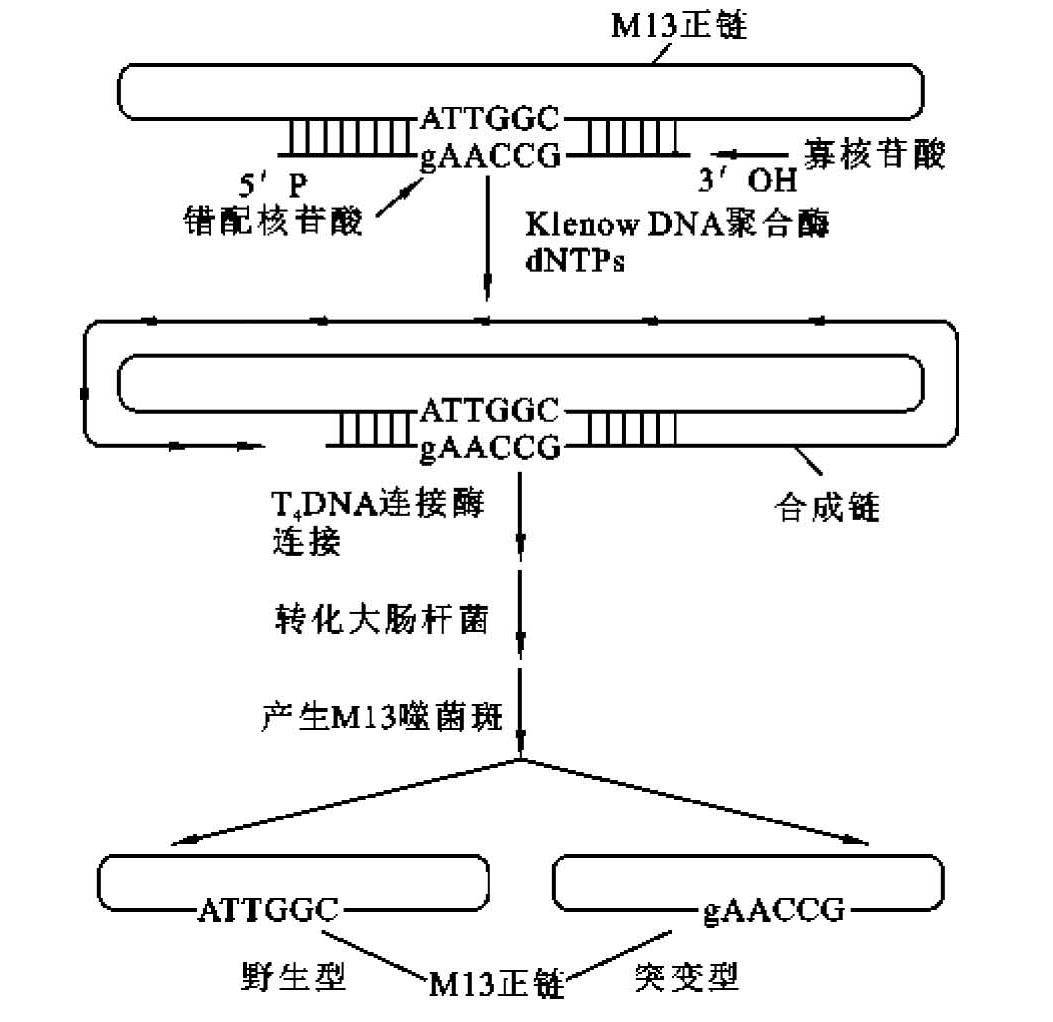
图13-7 寡核苷酸诱变过程示意图
(引自刘贤锡,2002)
3)Kunkel定点诱变法
体外DNA合成往往是不完全的,所以部分合成的DNA分子必须通过蔗糖密度梯度离心法除去。理论上来说,DNA是半保留复制的,应用寡核苷酸定点诱变时,所形成的噬菌体中携带突变基因的应为一半。但实际上由于宿主错配修复的反选择及其他技术上的原因,通常只有1%~5%的噬菌斑含有突变基因的噬菌体。因此,为了获得更多含有突变噬菌体的噬菌斑,必须提高突变体的比率。目前已有多种改良的寡核苷酸定点诱变方法,此处将简单介绍Kunkel在1985年建立的方法。
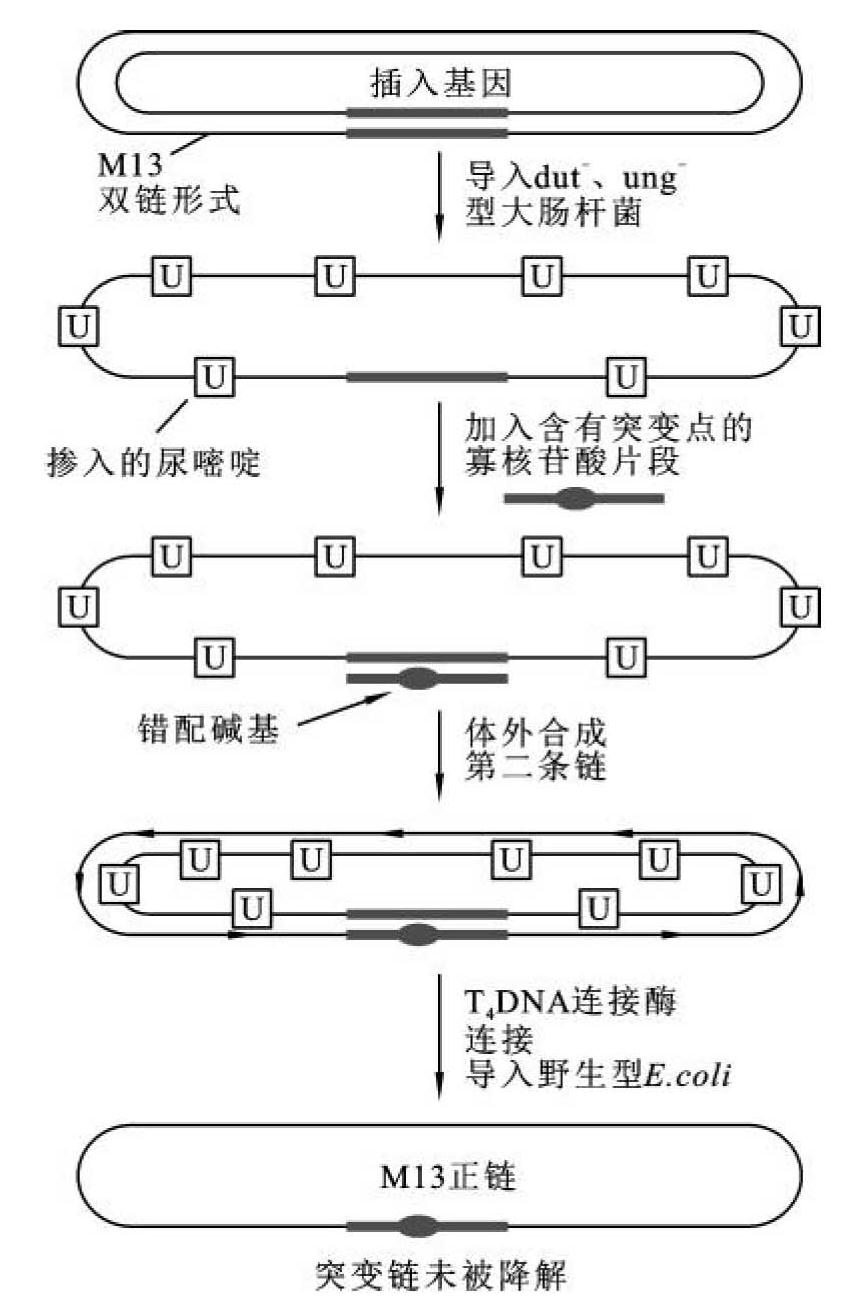
图13-8 Kunkel定点诱变示意图
(引自刘贤锡,2002)
Kunkel定点诱变法是一种通过筛除含有尿嘧啶的DNA模板链进行的寡核苷酸定点诱变法。它的基本原理是,将待突变的基因克隆入RF-M13 DNA载体上,导入具有dUTP酶(dut)和N-尿嘧啶脱糖苷酶(ung)双缺陷的大肠杆菌(dut-、ung-)菌株中。dut缺陷导致细胞内dUTP水平上升,并在DNA复制时,部分取代dTTP进入DNA新生链中。又由于ung缺陷,掺入DNA的dUTP残基不能除去。由这种大肠杆菌菌株产生的M13单链DNA大约有1%的T被U取代,以此为模板链,利用含有突变位点的寡核苷酸片段引导合成互补链。双链DNA导入正常的大肠杆菌中,其尿嘧啶N-糖基化酶除去DNA链上的尿嘧啶碱基。结果原来的M13模板链被降解。只有突变链因不含U,被保留下来(图13-8),这种方法产生的M13噬菌体中含有突变DNA的比率大大增加。
2.PCR定点诱变
寡核苷酸诱变方法所使用的M13噬菌体系统,操作起来比较烦琐,诱变周期较长。随着PCR技术的不断发展,人们将其应用在诱变技术中,使得此技术得以简化。
PCR定点诱变又称为PCR寡核苷酸定点诱变,该法具有简单、快捷的特点,其基本操作程序(图13-9)如下。①将待诱变靶基因克隆到质粒载体上,并分装到两支反应管中。②在每一支反应管中加入两种特定的引物,其中引物1和3均含有错配核苷酸,但两种引物分别与质粒DNA的不同链不完全互补,引物2和4均不含错配核苷酸,两者分别与质粒DNA的引物1和2杂交链的互补链完全互补。③进行PCR扩增获得含有突变碱基的线形质粒DNA。④将两支反应管中的线形质粒DNA混合,再经过变性和复性,一支反应管中的一条链和另一支反应管中的互补链杂交,通过两个黏性末端形成带有缺口的环状DNA分子。⑤转化大肠杆菌,环状DNA分子的缺口可被大肠杆菌修复。如果同一支反应管中的两个互补链又互相杂交,则继续形成线状DNA分子,在大肠杆菌中不稳定,易被降解。该方法把特异突变点导入克隆基因,无须把基因插入M13中,即可在大肠杆菌中进行表达。
3.盒式诱变
盒式诱变(cassette mutagenesis)是一种定点诱变技术,其方法是利用一段人工合成的具有突变序列的双链寡核苷酸片段,取代野生型基因中相应序列(图13-10),这种诱变的双链寡核苷酸片段是由两条人工合成的寡核苷酸链组成的,当它们退火时,会按照设计要求产生出克隆需要的黏性末端。这些合成的寡核苷酸片段就好像不同的盒式录音磁带,可随时插入已制备好的载体分子(“录音机”)上,便可以获得数量众多的突变体,故称为盒式诱变。该方法的优点是简单易行,突变效率高。
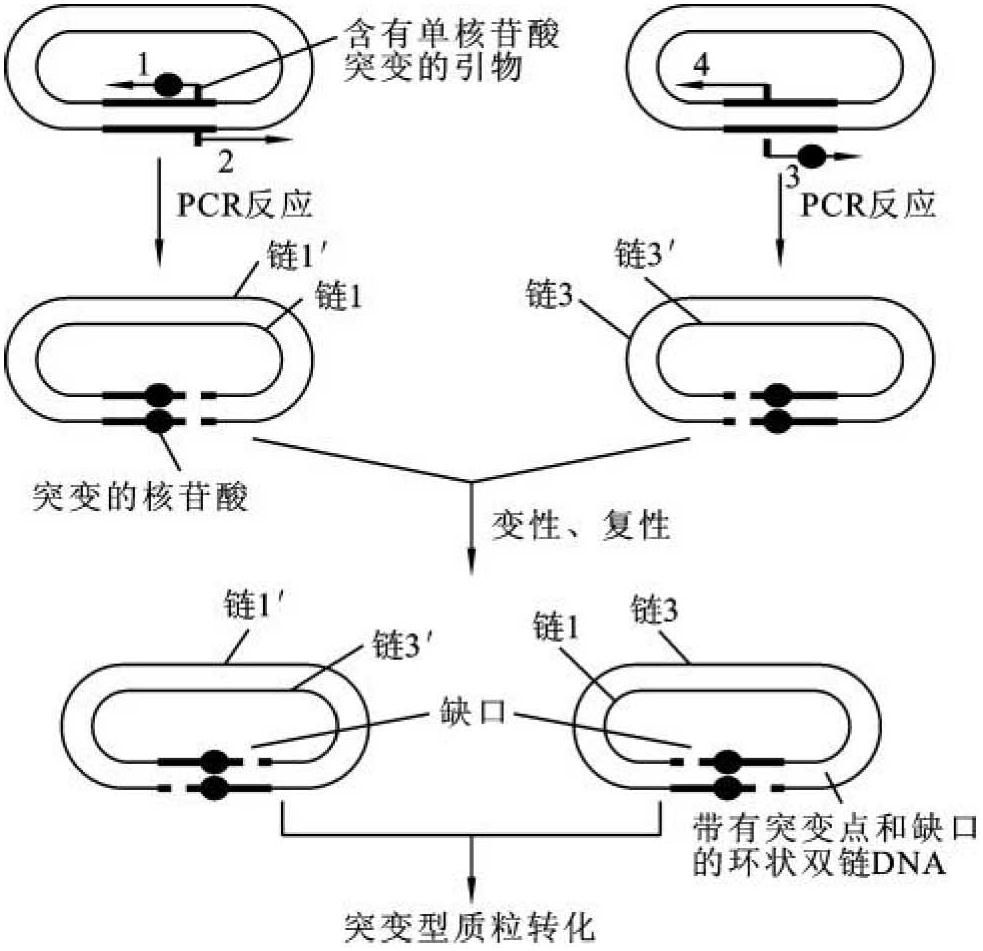
图13-9 PCR进行寡核苷酸定点诱变的示意图
(引自刘贤锡,2002)
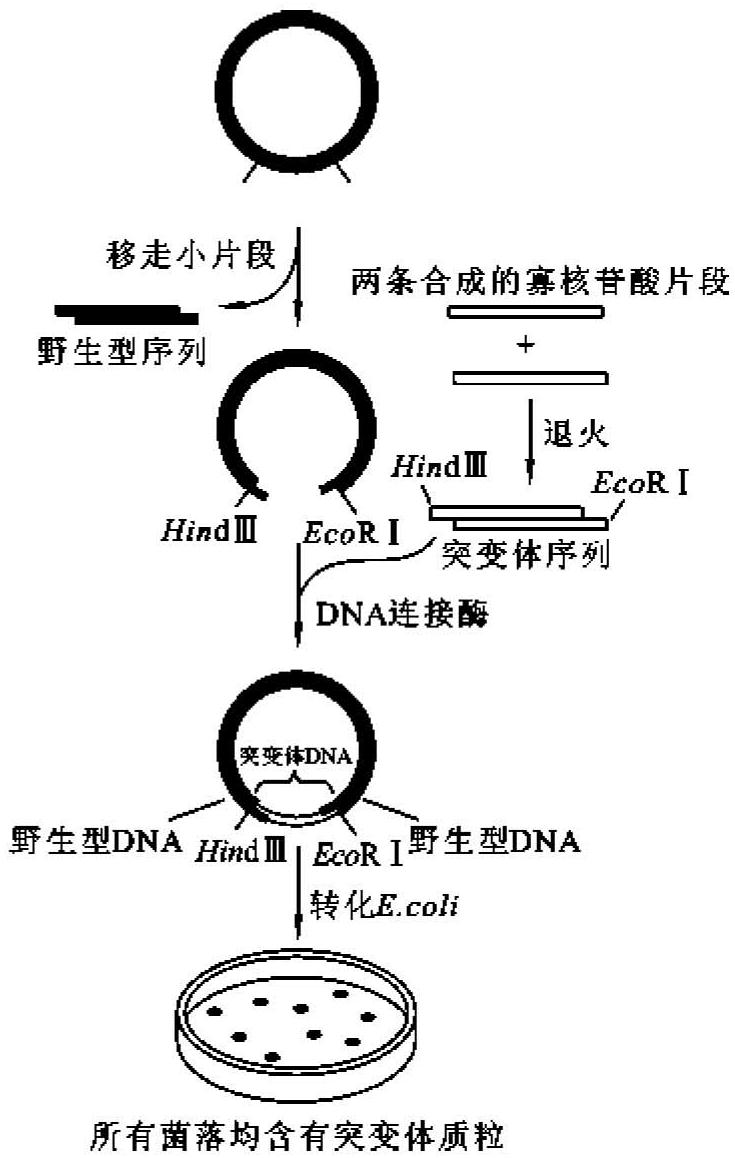
图13-10 盒式诱变示意图
(引自刘贤锡,2002)
4.随机诱变
定位诱变通常需要了解目的序列的详细情况,当缺乏这方面的资料和信息时,定位诱变方法的利用就受到限制,在这种情况下,利用随机诱变方法,在目的序列中产生突变,仍可以用于研究目的蛋白或目的核酸序列的结构和功能。
随机诱变(random mutagenesis)的工作原理(图13-11)是,将待突变基因克隆到一个载体的特定位点上,其下游紧接着是两个限制性核酸内切酶的酶切位点(RE1、RE2),前一酶切位点是5'突出(3'凹陷)的单链末端,后一酶切位点是3'突出(5'凹陷)的单链末端。然后用大肠杆菌核酸外酶(exonuclease Ⅲ,Exo Ⅲ)处理酶切缺口。ExoⅢ的主要活性是催化双链DNA自3'-羟基端逐一释放5'-单核苷酸,其底物是线状双链DNA或有缺口的环状DNA,而不能降解单链或双链DNA的3'突出末端。因此,当用ExoⅢ处理时,则可逐一水解3'凹陷末端。在适当时机,终止ExoⅢ的酶切反应,缺口用Klenow DNA聚合酶补平,底物为四种dNTP,再加上一种脱氧核苷酸的类似物。在缺口填补过程中,这个类似物会掺入DNA链上的一处或多处。再用S1核酸酶处理单链末端,形成平头末端,并用T4DNA连接酶连接。这种重组质粒转化大肠杆菌后,50%的基因上携带错配的碱基,导致位点变异。随机诱变的缺点是必须检测每个克隆,看哪一个产生了具有期望特性的蛋白质。这种检测不是一个简单的工作,但这是发现有新特性蛋白质的唯一方法。一旦发现了有潜在优点的突变体,通过确定哪个位点突变了可以确定克隆基因的序列。
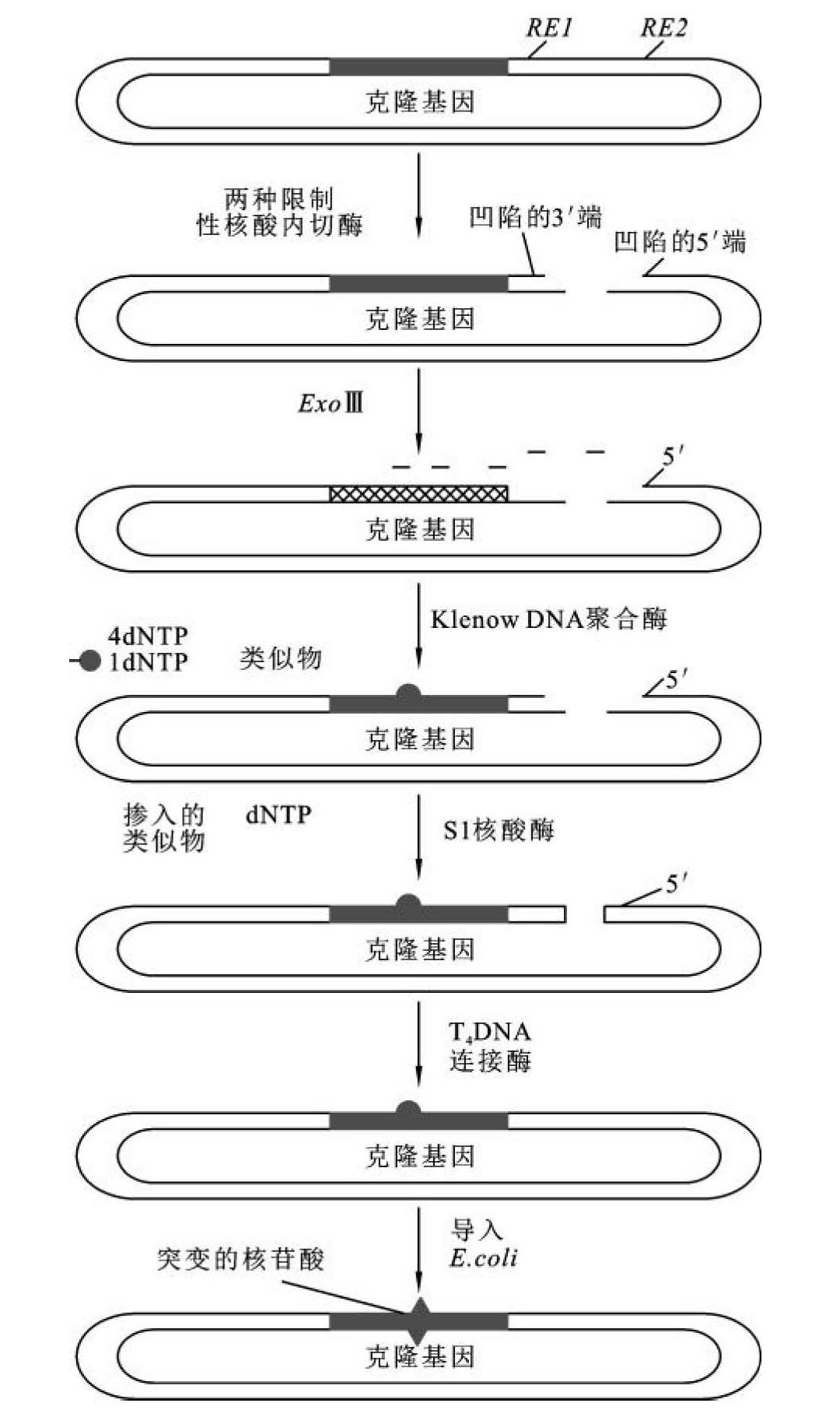
图13-11 随机诱变示意图
(引自刘贤锡,2002)
13.2.2 定向进化
酶的体外定向进化(directed evolution of enzyme in vitro)是改造酶蛋白质分子的一种新策略。它不需事先了解酶的定向结构和催化机制,在实验室模拟自然进化机制,通过由易错PCR、致突变菌株诱变等方法对编码酶蛋白质的基因进行随机诱变,由DNA改组(DNA shuffling)、随机引发重组和交错延伸等方法进行突变基因体外重组,设计高通量筛选方法来选出需要的突变株。它不仅可快速生产工业上有用的新酶,而且为研究蛋白质的结构与功能的关系开辟了崭新的途径,极大地拓展了蛋白质工程学的研究和应用范围。酶的体外定向进化的基本原理是,在待进化酶基因的PCR扩增反应中,利用Taq DNA聚合酶不具有3'→5'校对功能的性质,配合适当条件,如降低一种dNTP的量等,以很低的比率向目的基因中随机引入突变,构建突变库,凭借定向的选择方法,选出所需性质的优化酶(或蛋白质),从而排除其他突变体。简言之,定向进化就是随机突变加选择,前者是人为引发的,后者虽相当于环境,但只作用于突变后的分子群,起着选择某一方向的进化而排除其他方向突变的作用,整个进化过程是在人为控制下进行的。
1.定向进化的基本思路
定向进化是在不了解蛋白质的结构信息情况下,根据人为设定的目标,制备具有特定性质和功能的特殊蛋白质。在制订定向进化研究方案时需要考虑以下几个关键问题:
(1)随机诱变方法是通过引进随机的碱基替换,进而筛选理想的突变体。一般有益突变的频率很低,绝大多数突变是有害的。当突变频率太高时,几乎无法筛选到有益突变体;突变频率也不能太低,否则未发生任何突变的野生型将占据突变群体的优势,也很难筛选到理想的突变体。一般认为,理想的突变频率为每个目的基因的碱基替换在1.5~5个。
(2)组成一种蛋白质分子的氨基酸经排列组合可产生巨大的顺序空间,即可获得数量极大的排列方式,其中绝大多数排列方式没有反应功能,尤其是我们想要的功能。因此在考虑实验方案时,最好是选择一个性状最接近人们期望的酶分子作为起点。
(3)要从大量的突变体库中获得理想的个体,最重要的问题是筛选方法,筛选方法越好,成功的可能性越大。因此,应建立有效且灵敏的选择方法,确保检测出由单一氨基酸取代而引起的功能变化。
2.定向进化的常规技术
(1)易错PCR:易错PCR(error-prone PCR)是指在扩增目的基因的同时引入碱基错配,导致目的基因随机突变,改变PCR的条件,通常降低一种dNTP的量(降至5%~10%),或加入dITP来代替减少的dNTP,即会使PCR易于出错,达到随机突变的目的。然而,经一次突变的基因很难获得满意的结果,由此发展出连续易错PCR(sequential error-prone PCR)策略。即将一次PCR扩增获得的有用突变基因作为下一次PCR扩增的模板,连续反复地进行随机诱变,使每一次获得的小突变累积而产生重要的有益突变。Chen等利用此策略定向进化枯草芽孢杆菌蛋白酶E的活性获得成功,所得突变体PC3在高浓度二甲基酰胺(DMF)中,酶催化效率是野生酶的256倍。将PC3再进行两个循环的定向进化,产生的突变体13M的催化效率比PC3高3倍。用易错PCR法进行定向进化改造,关键在于突变率的控制。另外,该法较为费力、耗时,一般适用于较小的基因片段(800 bp以下)。
(2)DNA改组:DNA改组(DNA shuffling)是Stemmer于1994年建立的模仿自然进化的一种DNA体外随机突变方法。所谓DNA改组,就是将DNA拆散后重排,即将一种基因或具有结构同源型的几种基因在DNaseⅠ的作用下随机酶切成小片段,这些小片段之间有部分的碱基序列重叠,它们通过自身引导PCR(self-priming PCR)延伸,并重新组装成全长的基因,这一过程称为再组装PCR(reassembly PCR)。DNA改组的基本过程包括四个步骤(图13-12)。①目的基因的准备:根据需要选择一个基因或其片段,也可以是几个序列上具有较高同源性的基因。②DNase Ⅰ酶切:将目的基因随机切割成10~50 bp或300 bp左右的小片段。③不加引物的PCR:在Taq DNA聚合酶的作用下将DNaseⅠ切割后的DNA重叠小片段重新连接起来,在此过程中可能发生许多突变和重组。④加入引物的PCR:加入目的基因片段两端的引物,使连接好的DNA得到扩增,筛选正突变重组子。得到的正突变重组子又可以重复进行改组,使性状进一步提高。DNA改组的目的是创造将目的基因群中的突变尽可能组合的机会,导致更大的变异,最终获取最佳突变组合酶。在理论和实际上,它都优于重复寡核苷酸引导的诱变和连续易错PCR。通过DNA改组,不仅可加速积累有益突变,而且可使酶的2个或更多的已优化性状合为一体。
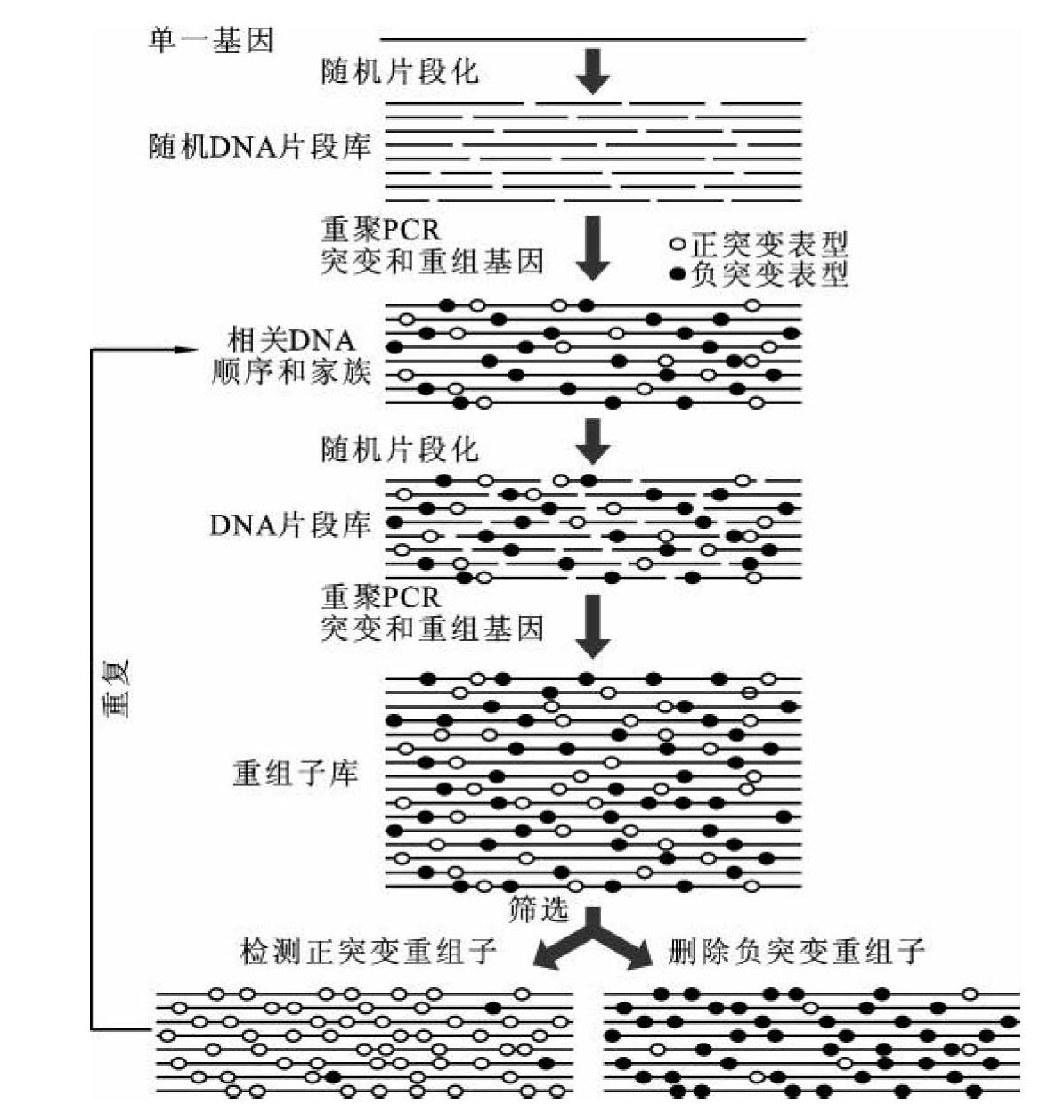
图13-12 DNA改组原图
(引自刘贤锡,2002)
(3)随机引发重组:随机引发重组(random-priming recombination,RPR)是Aronld于1998年首先报道的。其基本原理是以单链DNA为模板,配合一套随机序列引物,先产生大量互补于模板不同位点的DNA小片段,由于碱基的错误掺入和错误引发,在随后的PCR中,它们互为引物进行合成,伴随重组,再组装成完整的基因(图13-13),克隆到表达载体上,随后筛选。该法同DNA改组相比有以下优点:①RPR直接利用单链DNA或mRNA为模板;②在DNA改组中,片段重新组装前必须彻底除去DNase Ⅰ,而RPR方法省去了DNase Ⅰ切割成片段的过程,因而使基因的重组更容易;③合成的随机引物具有同样长度,无顺序倾向性,保证了点突变和交换在全长的后代基因中的随机性;④随机引发的DNA合成不受DNA模板长度的限制。
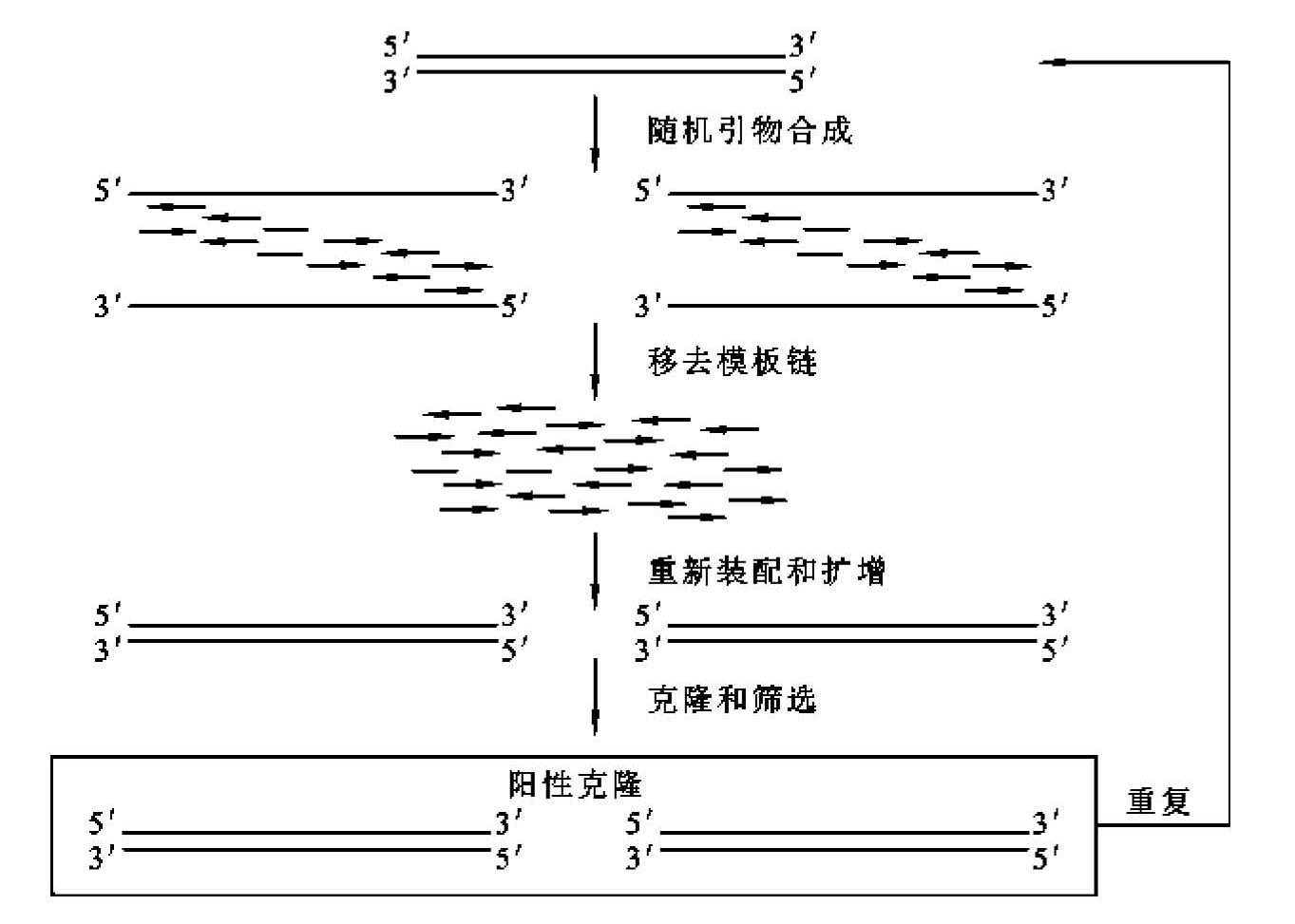
图13-13 体外随机引发重组原理
(引自刘贤锡,2002)
(4)交错延伸技术:交错延伸(stagger extension process,StEP)是Aronld等于1998年建立的一种新的体外重组方法。其原理是,在PCR中把常规的退火和延伸合并为一步,并缩短其反应时间(55℃,5s),从而只能合成出非常短的新生链,经变性的新生链再作为引物与反应体系内同时存在的不同模板退火并延伸。此过程重复进行,直到产生完整的基因长度,结果产生间隔的与不同模板序列互补的新生DNA分子(图13-14)。该技术已成功地重组了由易错PCR产生的5个热稳定的枯草芽孢杆菌蛋白酶E的突变体,得到热稳定性进一步提高的重组酶。
交错延伸法重组在单一试管中进行,不需分离亲本DNA和产生的重组DNA。它采用的是变换模板机制,这正是逆转录病毒所采用的进化过程。该法简便、有效,为酶的体外定向进化提供了又一强有力的工具。
13.2.3 tRNA介导的蛋白质改造
tRNA主要起转运氨基酸的作用。由于tRNA分子具有同工性,即一种以上的tRNA对应于一种特异氨基酸,因此细胞内tRNA的种类(80多种)比氨基酸的种类多。tRNA介导的蛋白质工程(tRNA-mediated protein engineering,TRAMPE)则允许拓展遗传密码,从而可以将人为设计的非天然氨基酸选择性地掺入蛋白质,产生具有新的或更好性状的蛋白质。利用TRAMPE进行的早期研究是基于对可以携带天然氨基酸的tRNA的化学修饰,现在称为非天然氨基酸替代(non-natural amino acid replacement,NAAR)法。该突变tRNA通过改变其反密码子能够识别并抑制终止密码子的链中止效应。这种方法允许蛋白质中特定的氨基酸由它们的衍生物来替代。随后,Noren等发展了定点非天然氨基酸替代(site-directed NAAR,SNAAR)的方法,在编码蛋白质的mRNA的特定位点上引入终止密码子,最后用互补的错氨酰化校正tRNA来通读越过此终止密码子,从而在蛋白质的特定位点掺入非天然氨基酸。利用天然氨基酸掺入技术,将特定的氨基酸掺入蛋白质肽链中的特定位点,这种修饰既能定性又能定量,同时一些传统修饰方法不能得到的衍生物,也可以通过该技术引入蛋白质的肽链之中。Sisido等列举了29种已经可以掺入蛋白质中的非天然氨基酸(图13-15)。
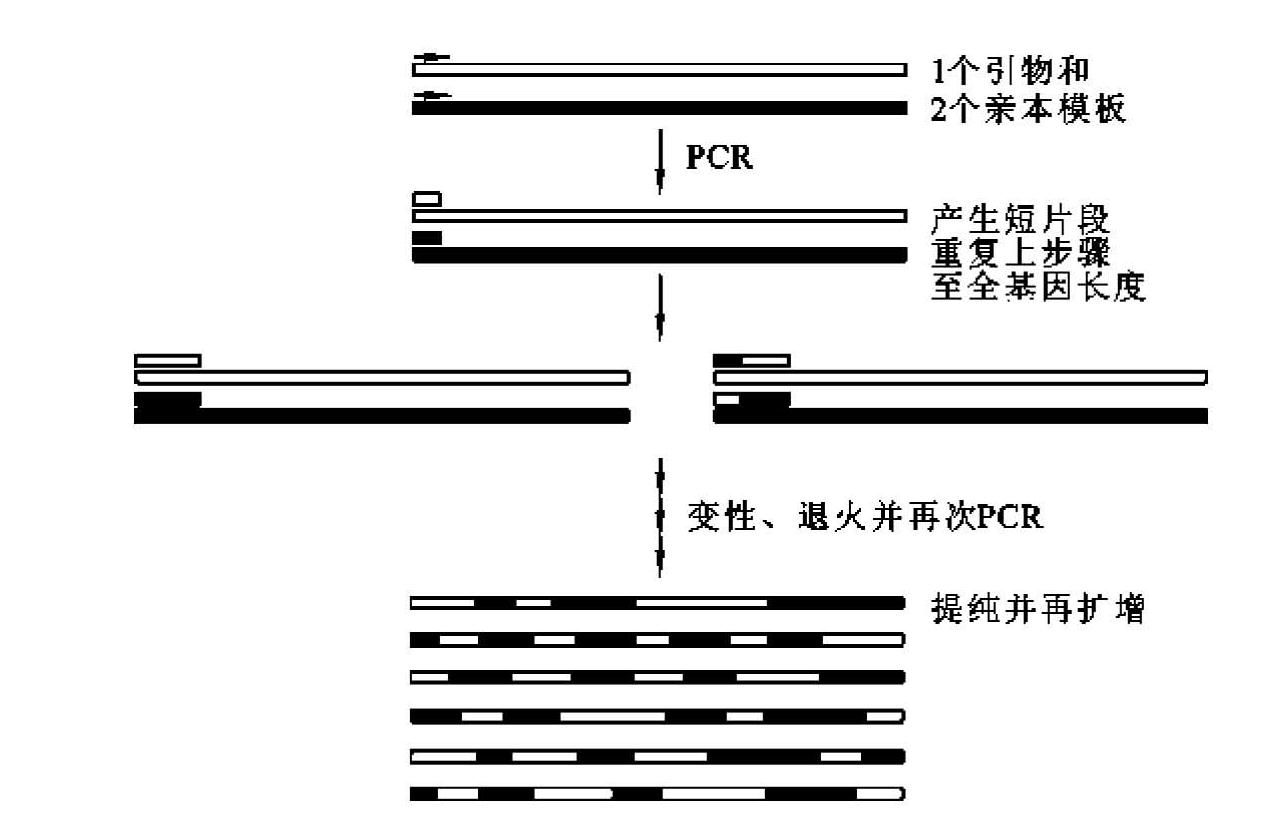
图13-14 交错延伸原理
(引自刘贤锡,2002)
生物体内共有20种氨酰tRNA合成酶,每一种均对应于一种天然氨基酸和一种tRNA类型,但是非天然氨基酸无相应的氨酰tRNA合成酶。目前已经发展了几种使tRNA被非天然氨基酸酰化的方法。
最简单的将非天然氨基酸和tRNA连接的方法是化学合成法。一种是用相应的氨酰tRNA合成酶氨酰化tRNA,使之携带天然的氨基酸,一旦共价键形成,即可以采用化学修饰的方法加入其他基团。第二种是化学法氨酰化,将非天然氨基酸氨酰化二核苷酸CA,然后通过T4 RNA连接酶连接到已去除3'端CA二核苷酸的tRNA上(图13-16(a))。两法相比较,酶促反应局限于天然的氨基酸及其类似物,而化学法可以氨酰化任何非天然的氨基酸。利用天然氨酰tRNA合成酶将一些氨基酸的类似物连接到原有tRNA是很少见的特例,但可改造原有的氨酰tRNA合成酶,使它们能识别一些非天然的氨基酸,而不再作用于天然的氨基酸(图13-16(b))。利用具有一定特异性核酶催化的转酶反应,将原来具有催化活性的RNA上的氨基酸转移到tRNA上(图13-16(c))。
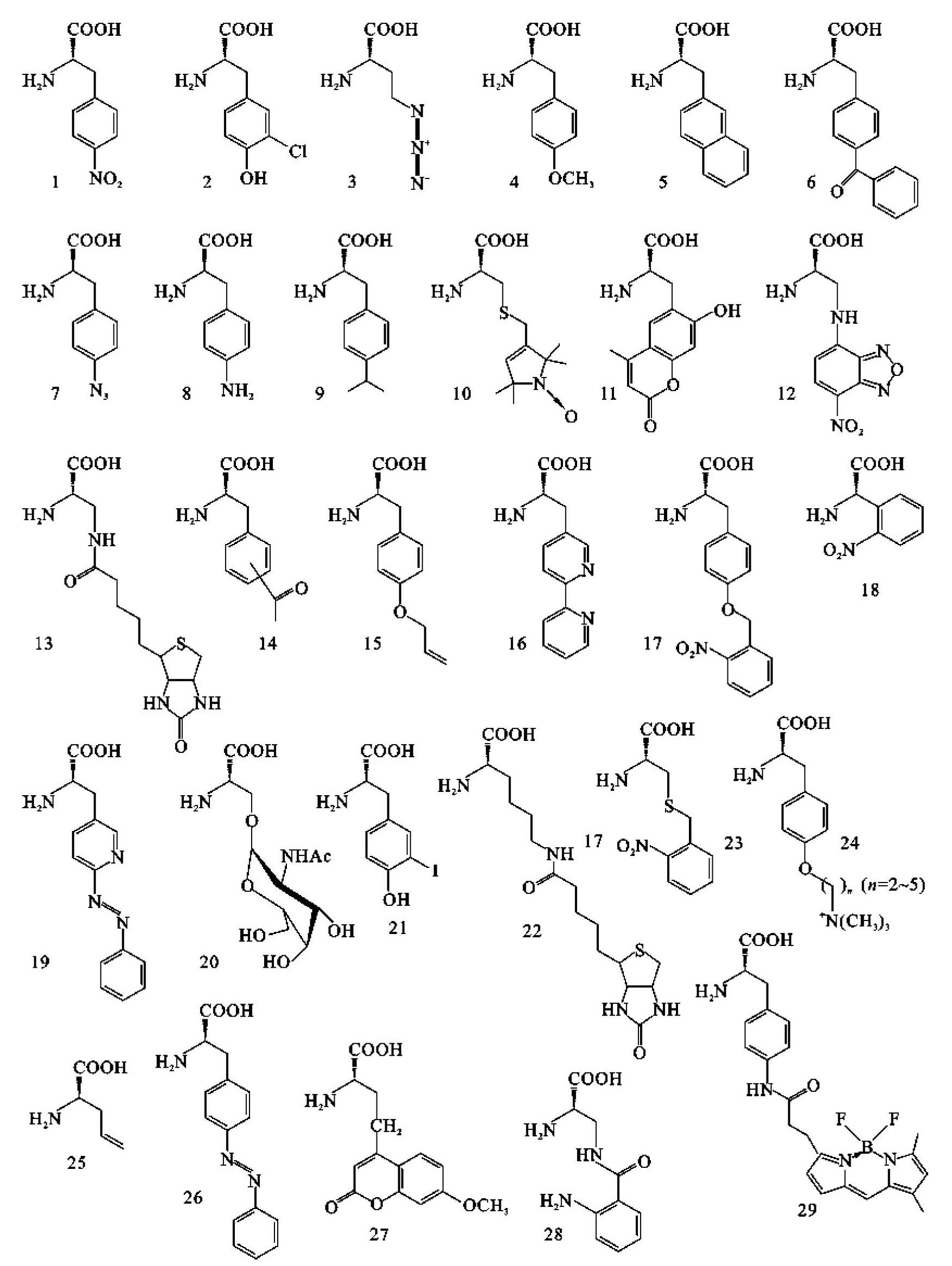
图13-15 通过tRNA介导掺入蛋白质中的非天然氨基酸
(引自梅乐和,2011)
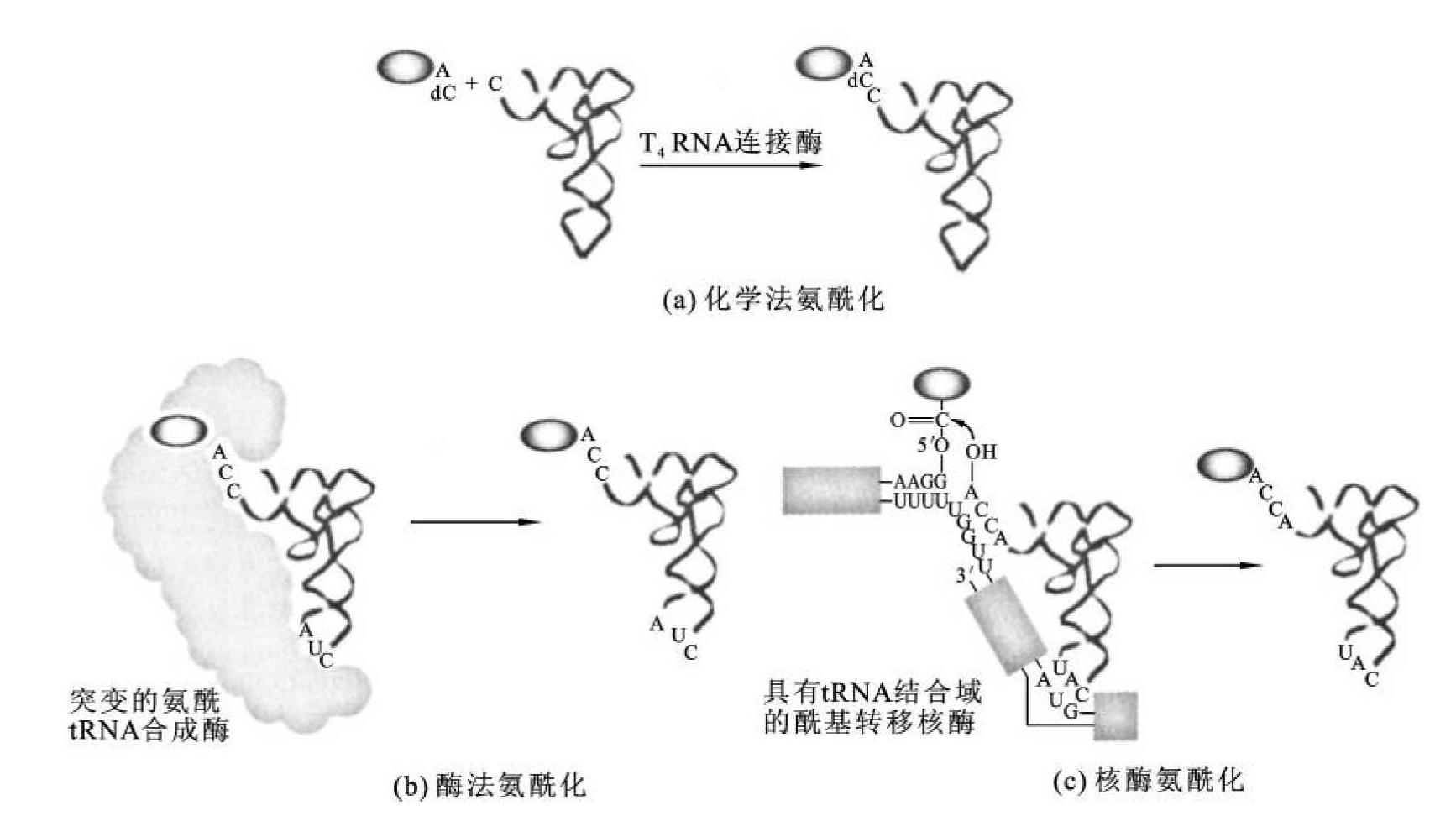
图13-16 非天然氨基酸和tRNA连接的方法
(引自梅乐和,2011)
13.3 蛋白质工程的设计思想
蛋白质分子设计,就是通过蛋白质结构的测定和分子模型的建立,按照蛋白质结构与功能的关系,综合运用各学科的技术手段,获得比天然蛋白质性能更优越的新型蛋白质。蛋白质分子设计探索蛋白质的折叠机理,为蛋白质工程提供指导信息。
13.3.1 蛋白质分子设计的分类
在蛋白质分子设计的实践中,常依据改造部位的多少将蛋白质分子设计分为三类。
1.定点突变或化学修饰法
这类蛋白质分子设计只进行小范围改造,是指在已知结构的天然蛋白质分子多肽链内的确定位置上,进行一个或少数几个氨基酸残基的改变或进行化学修饰,以研究和改善蛋白质的性质和功能,也称为“小改”。置换、删除或插入氨基酸,现在主要是通过分子生物技术从基因水平上操作来实现的。这一类蛋白质分子设计是目前蛋白质工程中最广泛使用的方法,主要是通过定点突变技术或盒式替换技术来有目的地改变几个氨基酸残基。
2.拼接组装设计法
这类蛋白质分子设计需进行较大程度的改造,是指对来源于不同蛋白质的结构域进行剪裁、拼接、组装,以期转移相应的功能,获得具有新特点的蛋白质分子,也称为“中改”。蛋白质的立体结构可以看作由结构元件组装而成的,因此可以在不同的蛋白质之间成段地替换结构元件。要进行蛋白质分子的剪裁,当前也主要是从基因水平上操作完成,故又称为“分子剪裁”。
3.从头设计全新蛋白质
这类蛋白质分子设计是从设计氨基酸序列结构开始进行全新蛋白质的设计,即从头设计一个全新的(自然界不存在的)蛋白质,使之具有特定的空间结构和预期的功能,也称为全新蛋白质设计或蛋白质从头设计。这类设计可参考已有的类似蛋白质三维结构,或完全依赖于已知的蛋白质结构与功能的知识,从基因水平上翻译表达出全新蛋白质,或直接进行化学合成全新的蛋白质。
13.3.2 蛋白质分子设计的原则
进行蛋白质分子设计需遵循一定的规则,以保证设计的成功。目前蛋白质分子设计还处于发展过程中,其规则还有待于继续完善。
(1)活性设计。活性设计是蛋白质分子设计的第一步,主要是考虑被研究的蛋白质功能,涉及选择化学基团和化学基团的空间取向。一般来讲,在这类设计中应采用天然存在的氨基酸来提供所需的化学基团,也可以通过加一些辅因子来达到预期的结果。
(2)对专一性的设计。专一性与酶的底物相关联。因此,酶蛋白化学基团与底物的专一性结合,对蛋白质分子设计十分重要。
(3)设计框架化。设计一个蛋白质活性分子必须框架化。在蛋白质活性分子中,该框架不仅可以提供各种活性基团的特定位置,而且还有其他功能,如吸附、运输以及可能的降解等。
(4)疏水基团与亲水基团需合理分布。因蛋白质分子的侧链并不总是完全亲水或完全疏水的,因此新设计的蛋白质分子要合理地分布亲水基团和疏水基团。要安排少量疏水残基在表面,少量亲水残基在内部,要在原子水平上区分侧链的亲水部分与疏水部分。
(5)最优的氨基酸侧链集合排列。蛋白质内部的密堆积表明侧链构象在折叠状态是一种能量最低的构象。蛋白质的侧链构象由两个空间因素决定,即旋转每条链的立体势垒和氨基酸在结构中的位置。为了获得蛋白质结构及功能的专一性,在构建一个蛋白质模型时必须满足所有适宜的空间要求,以及蛋白质折叠的空间限制条件。
13.3.3 蛋白质分子设计程序
蛋白质分子设计是一门试验性科学,是理论设计过程与试验过程相互结合的产物。在这个过程中,计算机模拟技术和基因操作技术是两个必不可少的工具。蛋白质分子设计的过程如下:首先通过生物信息学对所研究对象的结构和功能信息进行收集分析,建立研究对象的结构模型,在此基础上进行结构-功能关系研究,对其功能相关的结构进行研究和预测,然后提出蛋白质结构改造的设计方案,再通过基因工程改造得到设计产物,并通过相关试验进行验证,根据验证结果进一步修正设计,并且往往要经过几次这样的循环才能获得成功。一般的蛋白质分子设计工作可以按照以下步骤进行。
(1)收集相关蛋白质的结构信息。收集待研究蛋白质一级结构、立体结构、功能结构域及与之相关的同源蛋白质等相关数据,为蛋白质分子设计提供依据。
(2)建立所研究蛋白质的结构模型。可通过蛋白质、X射线、晶体学及NMR等方法测定蛋白质的三维结构。也可依据同源性较高蛋白质的三维结构,结合三维结构预测方法对待研究蛋白质进行结构预测。预测出的三维结构作为分子设计中的结构模型。
(3)进行结构模型的生物信息分析。分析其三维结构的特点、功能活性区以及结构中存在的二硫键数目和位置等,为选择设计目标提供依据。
(4)选择设计目标。确定所要建造的三级结构。考虑所要求的性质受哪些因素的影响,然后对各因素逐一进行分析。
(5)进行序列设计。充分考虑氨基酸残基形成特定二级结构的倾向性。选择和排布残基对时要考虑到疏水相互作用、螺旋的偶极稳定作用、静电相互作用、氢键作用以及残基侧链的空间堆积。尽可能地使序列有利于形成预期的二级结构。
(6)通过理论预测方法预测出所设计的多肽的二级结构和三级结构,初步检验设计的正确程度,检验目标模型与预期目标的吻合程度,并在此基础上进行调整。
(7)获得新蛋白质。对蛋白质分子进行计算机理论设计之后,进行实验室合成,同时也可通过基因工程手段人为合成或改造,然后进行基因表达,并分离纯化,获得新蛋白质分子。
思考题
1.名词解释:蛋白质工程;基因定点诱变;盒式诱变;易错PCR;生物信息学。
2.列表总结基因工程和蛋白质工程的相同点、不同之处(从结果、实质、流程等方面)及其联系。
3.蛋白质工程的基本目标、基本途径是什么?
4.简述蛋白质三维结构的预测方法。
5.假设待预测三维结构的目的蛋白质为U(un-known),简述利用同源模型化方法建立结构模型的过程。
6.简述定点诱变的方法。
7.简述M13寡核苷酸诱变过程。
8.简述Kunkel定点诱变法基本原理。
9.简述PCR定点诱变的操作程序。

扫码做习题
参考文献
[1]刘贤锡.蛋白质工程原理与技术[M].济南:山东大学出版社,2002.
[2]张惠展.基因工程[M].4版.上海:华东理工大学出版社,2017.
[3]黄迎春.蛋白质工程简明教程[M].北京:中国水利水电出版社,2009.
[4]梅乐和.蛋白质化学与蛋白质工程基础[M].北京:化学工业出版社,2011.
[5]唐建国,茹炳根,徐长法,等.蛋白质工程的研究[J].北京大学学报(自然科学版),1998,34(2-3):342-349.
[6]汪世华.蛋白质工程[M].北京:科学出版社,2008.