11.4.3 CRISPR-Cas系统
1.概述
CRISPR(clustered regularly interspaced short palindromic repeats)指有规律的成簇间隔短回文重复序列,Cas(CRISPR-associated proteins)指CRISPR关联蛋白。CRISPR-Cas系统是来源于细菌和古细菌,在抵抗入侵的病毒和外源DNA的长期演化中形成的一种适应性免疫防御。1987年,日本大阪大学实验室对K12大肠杆菌的碱性磷酸酶基因进行研究时,发现在该基因的编码区附近存在24~37 bp的串联间隔重复序列,但当时并没有引起普遍关注。后来,西班牙科学家在地中海盐菌中也发现了相同的序列。随后的研究发现这种间隔重复序列广泛存在于细菌和古细菌的基因组中。2002年,正式将这样的一段带有间隔序列的具有回文结构的多个重复序列命名为CRISPR。2005年,三个研究团队均发现CRISPR的间隔序列与宿主菌的染色体外的遗传物质高度同源,推测可能是微生物适应性免疫系统的一部分。2007年,Barrangou等首次证明在细菌遭受噬菌体的攻击后,在筛选得到的抗性细菌的CRISPR区整合了来源于噬菌体DNA的新的间隔序列,细菌通过识别与噬菌体序列相同的CRISPR间隔区对应的特定序列,抵抗噬菌体的入侵,获得对噬菌体的抗性,产生适应性免疫能力,抗性的特异性由噬菌体的间隔序列决定。2008年,John van der Oost等发现细菌CRISPR可转录并加工非编码RNA,即crRNA,而crRNA介导了随后的干扰机制。2011年,法国科学家在《Nature》上进一步阐明了CRISPR介导的免疫机制。2013年,张峰等率先实现了CRISPR-Cas9系统在哺乳动物细胞中特定位点的基因编辑,并构建了可同时靶向多个位点的基因编辑系统。从此,CRISPR-Cas基因编辑技术快速发展并得到广泛的应用。
2.CRISPR-Cas系统的作用机理
目前,CRISPR-Cas系统主要有五种类型,其中Ⅰ、Ⅲ和Ⅳ型需要多个Cas蛋白形成复合物才能干扰目的基因,但Ⅱ和Ⅴ型只需要利用单一Cas蛋白就能够干扰靶基因。目前,研究较透彻、应用较广的CRISPR-Cas9系统为Ⅱ型CRISPR系统,而Ⅴ型CRISPR系统就是新兴的CRISPR-Cas12a(Cpfl)系统。
CRISPR-Cas系统由CRISPR和Cas核酸酶组成(图11-7)。CRISPR基因座主要由前导区(leader)、重复序列(repeat)和间隔序列(spacer)构成。前导序列富含AT碱基,长度为300~500 bp,位于CRISPR基因上游,负责转录合成crRNA前体(Pre-crRNA)。重复序列长度为20~50 bp,并且涵盖5~7bp回文序列,转录产物可以形成发卡结构,稳定RNA的整体二级结构。间隔序列是被细菌捕获的外源DNA序列。当这些外源遗传物质再次入侵时,CRISPR-Cas9系统就会对目的序列进行切割,破坏外源DNA结构以达到防御的目的。
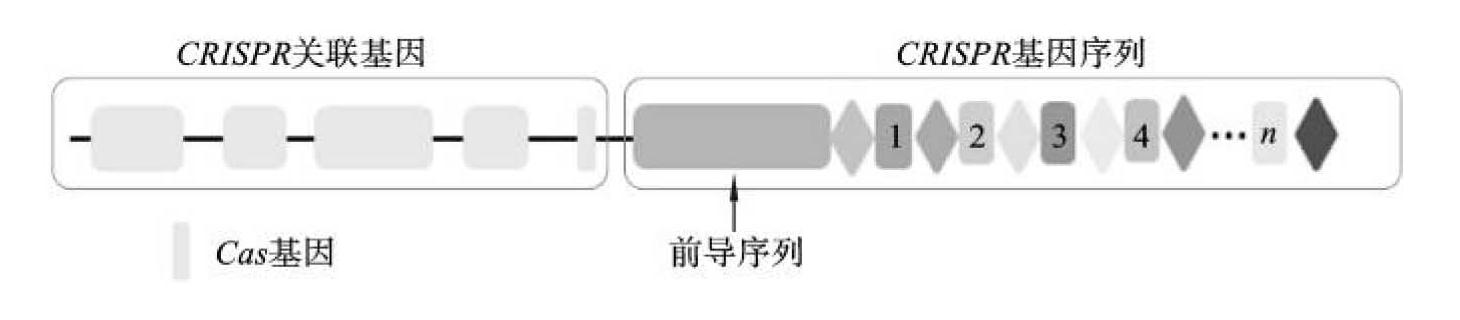
图11-7 CRISPR-Cas基因位点结构图
◆重复序列 n间隔序列
Cas基因位于CRISPR基因附近或分散于基因组其他地方,该基因编码的蛋白均可与CRISPR序列区域共同发生作用。因此,该基因被命名为CRISPR关联基因(CRISPR associated gene,Cas)。
CRISPR-Cas系统的作用机理可以分为三个阶段(图11-8)。第一阶段是CRISPR的新间隔序列的获得,外源DNA首次入侵时,细菌进入适应阶段,Cas蛋白会靶向裂解噬菌体上短基因片段(原间隔序列),将其插入宿主CRISPR基因座位点,这样使得CRISPR基因座中存在此种噬菌体的序列信息来形成新的间隔序列。第二阶段是crRNA(CRISPR-derived RNA)的成熟,即CRIPSR基因座的表达,包括转录和转录后的成熟加工。外源DNA再次入侵时,细菌激活了表达阶段,CRISPR基因座转录出前体crRNA,由内切核糖核酸酶催化加工成成熟的crRNA。第三阶段是CRISPR-Cas对外源遗传物质的识别与切割,也就是干扰阶段。成熟的crRNA会与tracrRNA(trans-activating crRNA,crRNA反式激活的RNA)形成双链结构,并与Cas蛋白形成复合体,激活Cas的核酸内切酶活性,识别并切割外源间隔序列中的PAM(proto-spacer adjacent motifs)位点,将外源DNA降解,从而抵抗同类噬菌体再次入侵,达到保护宿主的目的。PAM序列是靶向DNA序列3'端长度为3 bp的核苷酸序列,碱基组成通常为NGG(N为任一碱基)。
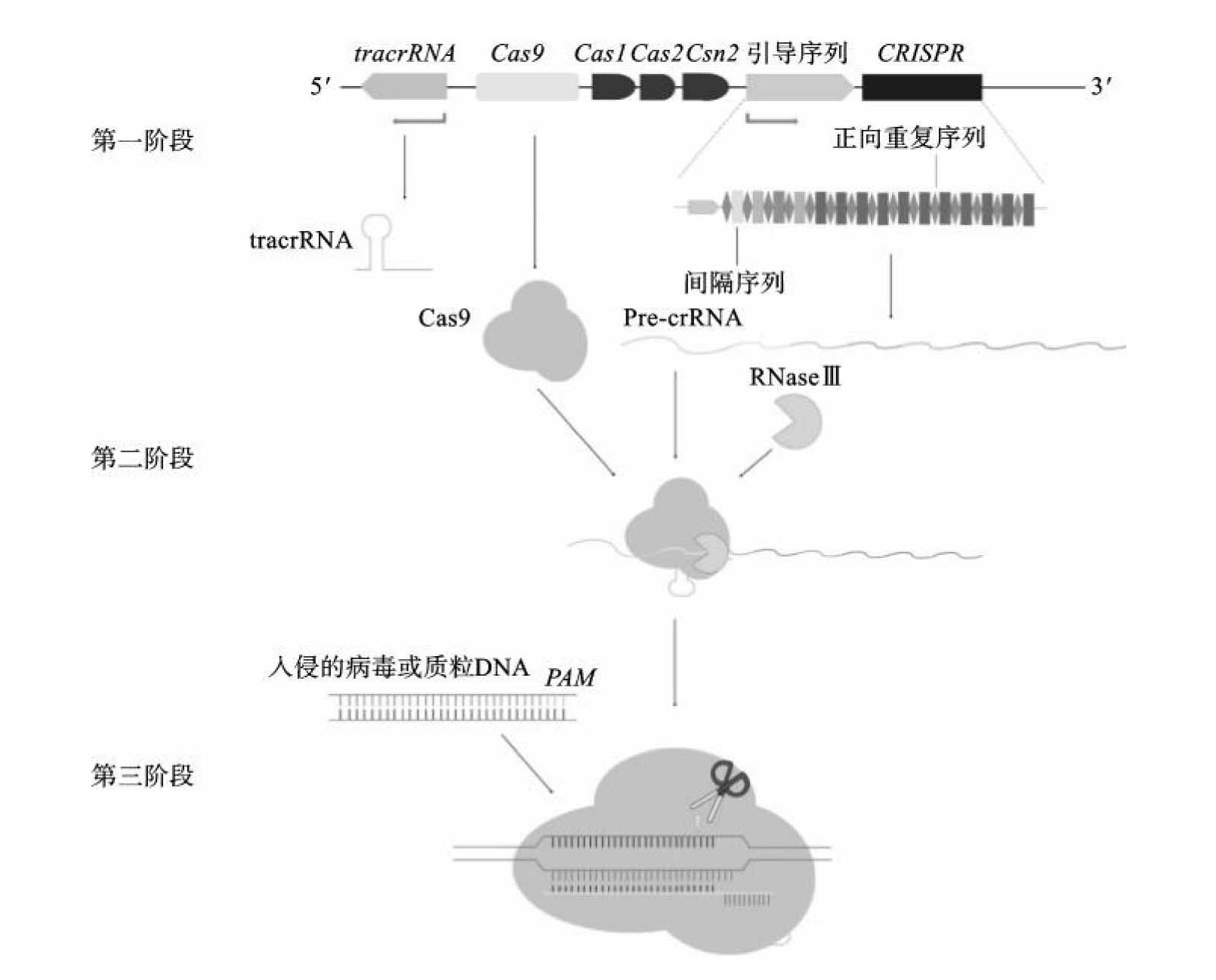
图11-8 产脓链球菌CRISPR-Cas9干扰噬菌体或外源质粒入侵示意图
3.CRISPR-Cas系统的建立
1)CRISPR-Cas9系统
基于Ⅱ型的CRISPR-Cas9系统,通过人工设计crRNA和tracrRNA,可以改造成具有引导作用的sgRNA(single guide RNA),引导Cas9蛋白在与crRNA配对的序列靶位点切割DNA双链(图11-9),这种全新的基因定点编辑技术——CRISPR-Cas9系统——已广泛应用于动植物研究中。
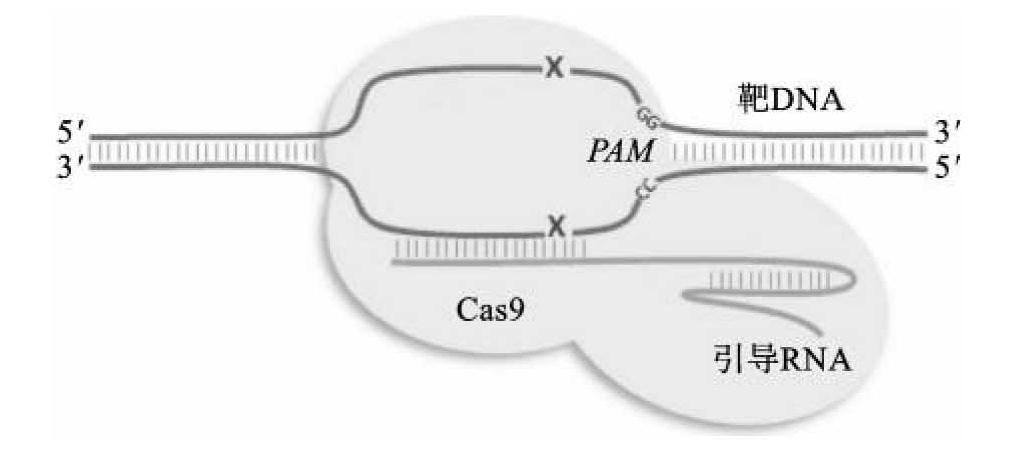
图11-9 人工设计的CRISPR-Cas9系统工作原理
CRISPR-Cas9系统主要包括两个方面:①构建Cas9-sgRNA表达载体,将载体导入受体细胞表达发挥编辑作用;②将表达纯化的Cas9蛋白与合成的sgRNA导入受体细胞发挥编辑作用。Cas9蛋白有2个重要的核酸酶结构域,即RuvC-like和HNH结构域,分别在靶DNA的PAM序列“NGG”上游3 nt处对DNA双链进行切割。由于Cas9来源于细菌,因此在真核系统中,需要在Cas9蛋白中添加一段核定位信号以保证该蛋白进入细胞核正常发挥功能。sgRNA是一段具有特定结构的单链RNA,其5'端约20 bp与靶DNA互补配对结合,引导Cas9-sgRNA复合物对相应位点进行切割,决定编辑位点特异性。
2)CRISPR-Cas12a(Cpfl)系统
尽管CRISPR-Cas9系统已广泛使用,但是该系统存在编辑位点受限制、容易脱靶等缺陷,因此,科学家们想建立一个新的CRISPR基因编辑系统。CRISPR-Cas12a系统属于V型CRISPR-Cas系统,该系统也只需一个Cas蛋白就能对双链DNA进行切割,但是相对于CRISPR-Cas9系统,CRISPR-Cas12a系统具有明显的特征和独特的优势(图11-10):第一,Cas12a仅携带RuvC-like结构域,不需要tracrRNA的参与,只需要在crRNA引导下即可切割双链DNA;第二,Cas12a特异性识别靶DNA富含T的PAM序列(5'-TTTN-3'或5'-TTTV-3'),然后在PAM序列下游对DNA双链进行切割,形成具有4~5 nt的黏性末端,增加了定向同源修复(homology-directed repair,HDR)途径发生的概率,有利于DNA片段更精准插入和替换;第三,CRISPR-Cas12a系统的crRNA比sgRNA更短,且Cas12a蛋白也比Cas9蛋白更小,因此,CRISPR-Cas12a系统适用于装载量小,特别是多靶点编辑的情况;第四,CRISPR-Cas12a系统在多个物种的基因编辑中均表现更低的脱靶率。但是,目前CRISPR-Cas12a系统仍局限于少数物种的基因编辑,这可能是因为该系统在低温条件下编辑效率低,并且对PAM序列要求较严格。
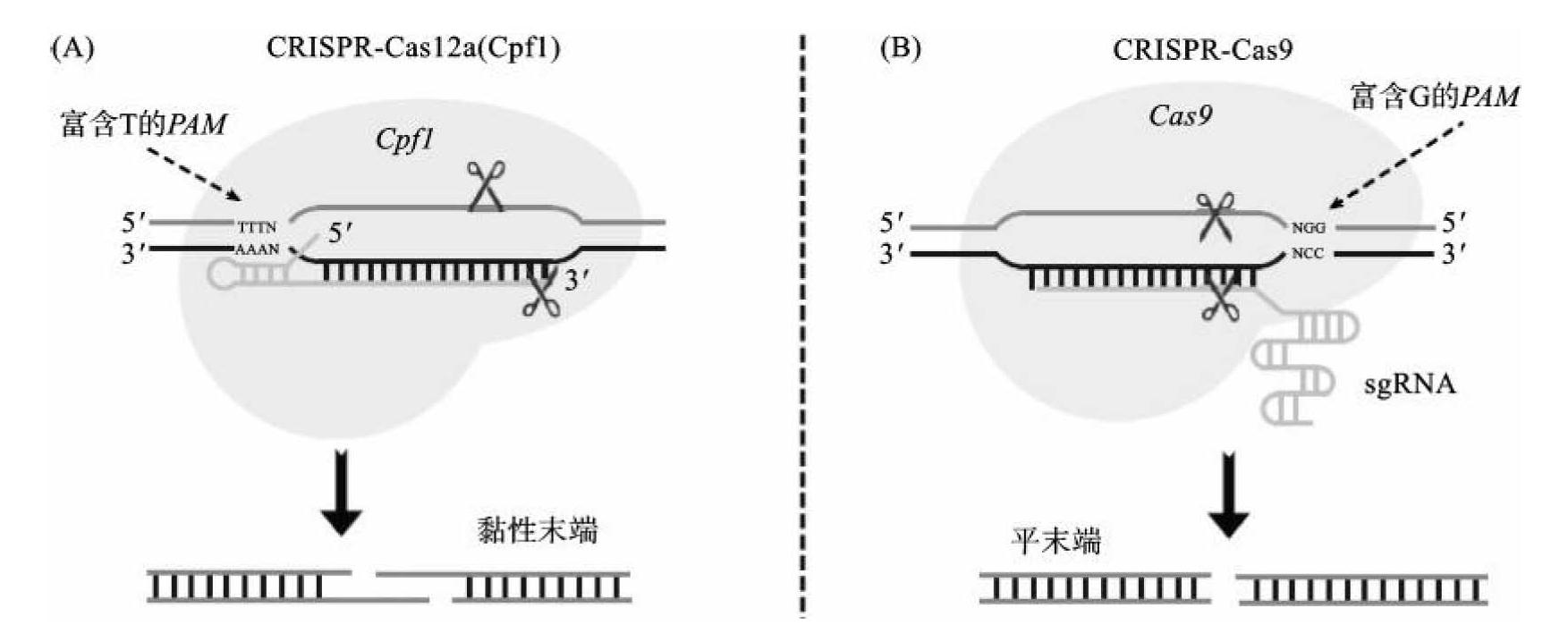
图11-10 CRISPR-Cas12a(Cpfl)系统与CRISPR-Cas9系统的比较
4.CRISPR-Cas基因编辑靶点的分析技术
CRISPR-Cas系统对靶DNA进行基因编辑,操作简单、突变率高,并且能实现对多个基因同时进行编辑。对于分析基因组编辑后的基因型,目前主要有三种靶点分析技术。(https://www.daowen.com)
1)利用Sanger测序的靶点分析方法
利用Sanger测序的靶点分析方法只适合于简单突变的样品。为了明确靶点基因序列的突变情况,经典的解码方法是设计基因位点特异引物,扩增包括靶点序列的DNA片段,克隆之后,挑取多个阳性克隆进行Sanger测序。这种方法耗时长并且昂贵。当突变类型为双等位突变和杂合突变时,测序图谱会出现延续杂乱的双峰。针对杂合突变和双等位突变,Ma等开发了一种叫简并序列解码(degenerate sequence decoding,DSD)的分析方法。DSD方法高效简单,并且能快速解码,但如果要分析较多的样品,这种手动解码还是效率太低。为了更好地解决这个问题,Liu等以DSD方法为原理编写程序,针对包含靶位点PCR扩增产物的测序文件直接解码,开发了一个基于网页的、多功能的直接解码工具DSDecode,可解码多种类型的突变,包括纯合突变、双等位突变、杂合突变等。为了快速处理大量的测序文件,Xie等进一步开发了一体化服务的软件包CRISPR-GE(http://sk1.scau.edu.cn),可对CRISPR-Cas9/Cas12a系统的靶点sgRNA进行设计,构建sgRNA载体引物,预测脱靶位点,对靶点突变位点解码等。因此,基因组编辑技术越来越自动化、人性化和简单高效。
2)利用高通量测序的靶点分析技术
当突变类型复杂,如一个靶点产生2个以上突变的嵌合突变,需要对多倍体物种的基因编辑进行解码,以及需要一次性对大量的靶点序列进行测序,针对这些情况,利用Sanger测序的靶点分析技术已不能满足需求。科学家们开发出利用高通量二代测序NGS(next generation sequencing)的靶点分析方法,如AGEseq、Cas-Analyzer、CRISPR-GA、CRISPResso和Hi-TOM等。Xue等开发了第一个用于分析NGS数据的基因编辑分析平台AGEseq(http://aspendb.uga.edu),它既是一个独立的程序,也是一个基于Galaxy的网页工具,该平台也可分析Sanger测序数据。Park等开发了一个基于JavaScript的NGS数据分析平台Cas-Analyzer,因为Cas-Analyzer完全是在客户端web浏览器上动态使用的,所以没有必要将非常大的NGS数据集上传到服务器,因而节省了大量的时间。该平台可分析各种基因编辑诱导的突变。Güell等开发了一个用于评估基因编辑质量的平台CRISPR-GA,操作简单,只需要点击3次鼠标就可完成评估过程。该平台可用于评估编辑位点的数量和突变效率,还能对插入、缺失位点,等位基因突变的效率等提供全面的报告。Pinello等开发了一个可定性和定量评估基因编辑效果的分析平台CRISPResso,可以评估序列的质量和比对的准确性,还有精确计算插入、缺失和碱基替换等多种功能。Liu等开发了一个可对多份样品和多个靶点的突变进行鉴定的平台工具Hi-TOM,特别适合通过基因编辑系统得到的所有类型突变的高通量鉴定,尤其是对复杂基因组编辑或复杂嵌合突变,具有很高的可靠性和灵敏度。
3)基于非测序手段的靶点分析方法
通过上述两种测序数据的方法分析靶点序列,可以直接获取突变的具体信息。但不通过测序,也可辨别基因编辑是否成功,比如PCR-RE(PCR/restriction enzyme)法、T7EⅠ(T7 endonucleaseⅠ)法和SSCP(single-strand conformational polymorphism)法等。PCR-RE法要求靶点处有特异的酶切位点,先用限制性核酸内切酶酶切基因组DNA,然后利用PCR扩增确认,限制性核酸内切酶的选择限制了该方法的使用。利用特异性切割错配分子的T7EⅠ或Surveyor酶也可以检测突变情况,其检测灵敏度较PCR-RE法低,但没有靶序列的限制。
5.CRISRP-Cas9系统的应用
近年来,CRISPR-Cas9系统广泛应用于各种生物研究领域,包括基因功能研究、遗传改良、构建动物模型等。基因编辑的方式主要体现在基因敲除、基因(片段)的定向插入和替换、单碱基编辑,以及基因表达调控等四个方面。
1)基因敲除
目前,利用CRISPR-Cas9系统对功能基因进行敲除,已广泛应用到各研究领域。通常CRISPR-Cas9系统会在切割位点附近产生碱基的插入、缺失,因而导致基因突变,还可通过CRISPR-Cas9系统同时进行多基因的编辑,甚至是大片段(1~100 kb)的删除。在动物的研究方面,Hwang等利用CRISPR-Cas9系统成功地在斑马鱼胚胎中实现了fhl、apoea等基因的定点突变;Wang等在小鼠中利用该技术实现了Tet1和Tet2等多个基因的同时定点突变。CRISPR-Cas9系统在植物的研究方面也取得了较多的成果。Li等利用CRISPR-Cas系统在模式植物拟南芥和本生烟中,对目的基因AtRACK1b、AtRACK1c和AtPDS3实现了基因组定点编辑,突变效率为1.1%~38.5%。Jiang等研究发现,在拟南芥T1代植株的体细胞中检测到很多Cas9-sgRNA介导的基因突变,这些突变可遗传到T2代和T3代中。Shan等利用CRISPR-Cas9系统对水稻OsPDS、OsMPK2以及OsBADH2基因进行定点的编辑,获得T0代OsPDS基因功能缺失的纯合突变体,呈现出矮小白化的表型。Zhou等利用CRISPR-Cas9系统将水稻中4个糖转运蛋白基因SWEET11、SWEET13、SWEET1a、SWEET1b进行编辑,在T0代转基因苗中目的基因的编辑效率达到87%~100%,同时能产生可遗传的长片段的缺失。
2)基因(片段)的定点插入或替换
通过CRISPR-Cas9系统进行基因编辑时,在DNA双链断裂的同时引入一个供体片段,并且在这个供体片段的两端设计与DNA断裂处相似的序列,此时编辑受体可能启动同源重组修复途径,通过同源重组实现供体片段的精确插入或替换。与在非同源末端连接(NHEJ)易错修复途径造成的随机插入或缺失相比,该编辑方式更加精准灵活,可实现多个基因的稳定聚合,解决了传统研究中无法连锁遗传的问题,因此具有更广泛的应用前景。
3)单碱基编辑
最初的单碱基编辑技术是依赖于胞嘧啶脱氨酶,对目的基因序列特定位点的单个碱基进行转换,也就是胞嘧啶编辑器(CBE),能实现C/G到T/A的转换。科学家不断研究又发现了腺嘌呤编辑器(ABE),可实现靶序列中A/T到G/C的转换。胞嘧啶编辑器和腺嘌呤编辑器系统能实现四种单碱基的编辑,已在水稻、小麦、玉米、番茄、拟南芥等多种植物中广泛应用。
4)基因的表达调控
利用CRISPR-Cas9系统调控基因表达主要有两种途径。一种途径是用Cas9蛋白对目的基因的启动子区的顺式调控元件(CRE)进行编辑,改变基因的表达水平或调控模式。Rodriguez-Leal等通过CRISPR-Cas9系统对番茄中多个基因的顺式调控元件进行编辑,获得了对QTL位点的突变,实现重要农艺性状的精准调控。另一种途径是将人工突变后的dCas9(Cas9核酸酶发生双突变,产生“钝化”和“死亡”的Cas9,即dead Cas9)蛋白与目的基因的转录调控结构域融合,然后通过sgRNA引导融合蛋白到目的基因的启动子区,抑制或激活该基因的表达。目前该方法在拟南芥、烟草和水稻等植物中都已成功应用。