9.4.5 外源基因整合位点分析
对于外源基因整合位点的精确分析则要通过分析外源基因插入位点的侧翼序列来完成。目前已知基因侧翼序列的分析方法有很多,比较有代表性的方法有质粒拯救法、热不对称交错PCR法、反向PCR法和高通量测序法。
1.质粒拯救法
1982年Holsters等首次将质粒拯救法(plasmid rescue)用于烟草基因组T-DNA插入位点侧翼序列的分离。其原理是构建T-DNA转化载体时,在左、右边界内部引入大肠杆菌的质粒复制起点及可在原核细胞中表达并起作用的选择标记基因;转化植物后,根据载体的序列选定T-DNA边界内合适的单一酶切位点对转化体基因组DNA进行完全酶切;酶切片段环化后转化大肠杆菌,含目的片段的环化载体可在大肠杆菌中进行复制从而存活于选择培养基上,筛选得到的阳性克隆经过测序可得到插入位点的侧翼序列信息(图9-20)。该方法在研究农杆菌介导转化整合情况时应用广泛。但对于常规意义的转基因作物育种,为避免带来生物安全性问题,在构建载体时不宜引入大肠杆菌的质粒复制起点及可在原核细胞中表达并起作用的选择标记基因。因此,对于准备推广应用的转基因作物不适合用该方法研究侧翼序列。
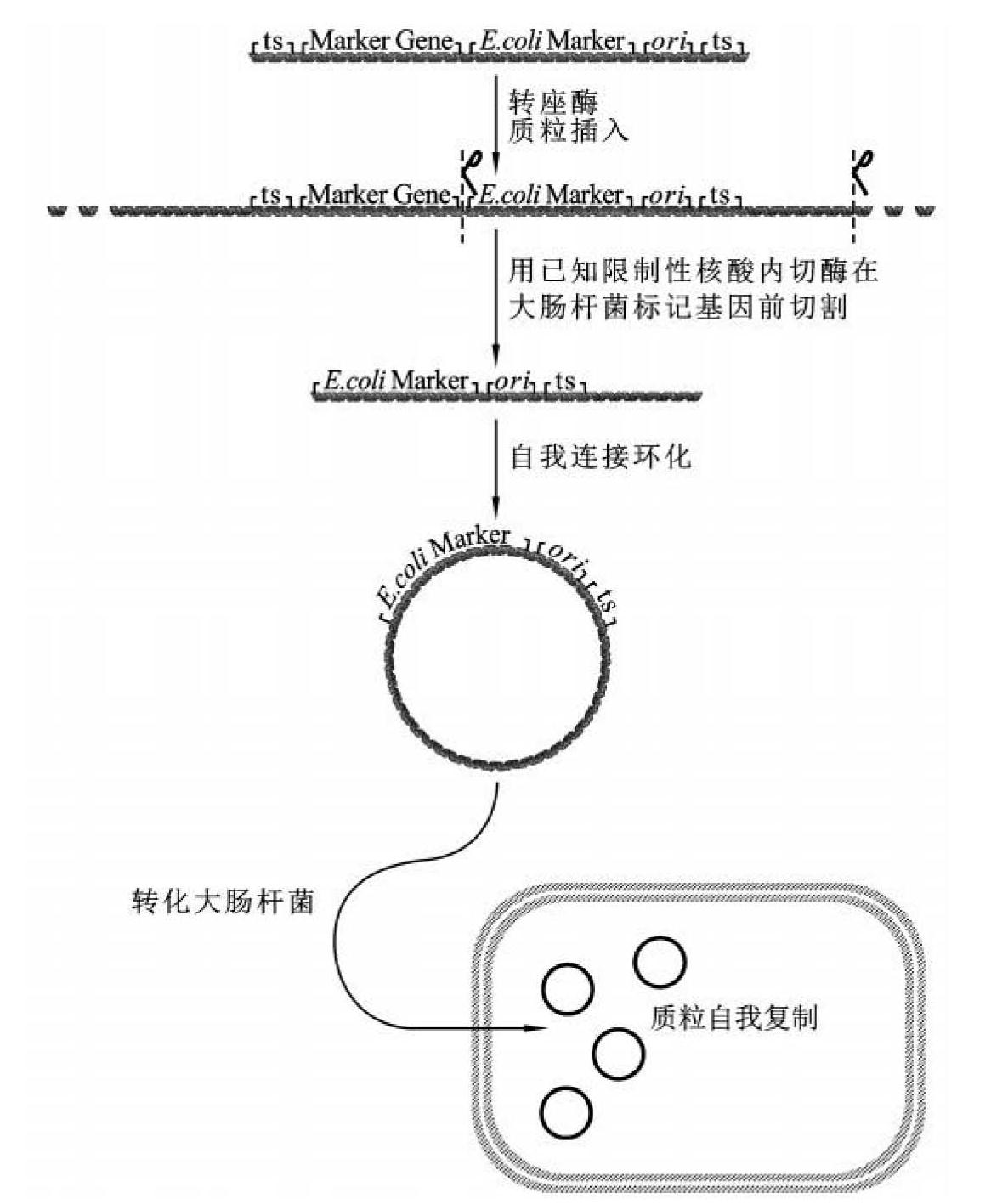
图9-20 质粒拯救分析整合位点侧翼序列程序
(引自Wikipedia)
2.热不对称交错PCR法
热不对称交错PCR法(thermal asymmetric interlaced PCR,TAIL-PCR)由Liu和Whittier在1995年创立。该方法的理论基础如下:依据已知序列设计的特异引物(TR)具有一个较高的Tm(解链温度)值,而设计的任意引物(arbitrary primer,AD)具有一个较低的Tm值,两者之间差值在10~15℃;在PCR过程中,使用几组热不对称交错PCR,即通过控制复性温度来调节特异引物和任意引物与模板的复性,实现对目的区域的特异性扩增(图9-21)。第一轮反应(primary reaction)是TAIL-PCR的重要环节,先进行5轮高严谨性循环,特异性引物(TR1)与模板退火,只能发生单引物循环,外源基因整合位点上游侧翼序列得到线性扩增。大幅度降低退火温度,使AD及TR1均与模板DNA相结合,指数式扩增一个循环。此后,两个高严谨性循环、一个低严谨性循环交替进行,共15个循环。特异性序列(两端分别拥有TR1和AD序列)和非特异性序列Ⅰ(只有TR1,没有AD序列)大大超过非特异性序列Ⅱ(两端均为AD序列)。PCR Ⅱ中,特异性序列再次被优先扩增,经稀释的非特异性序列Ⅰ也已大为降低,此时已没有明显的背景片段了。PCR Ⅲ是真正意义上的PCR,共20个循环,进一步扩增特异性序列,在第三阶段结束后,基本上只有目的产物。整个过程操作简单,容易实现自动化和大规模化分析。Liu和Whittier认为与以前的其他方法相比,TAIL-PCR具有7个很明显的优点:简单性、高特异性、高效性、高速性、无连接导致的假阳性、产物直接测序和高敏感性。TAIL-PCR一经报道,就得到极为广泛的应用。
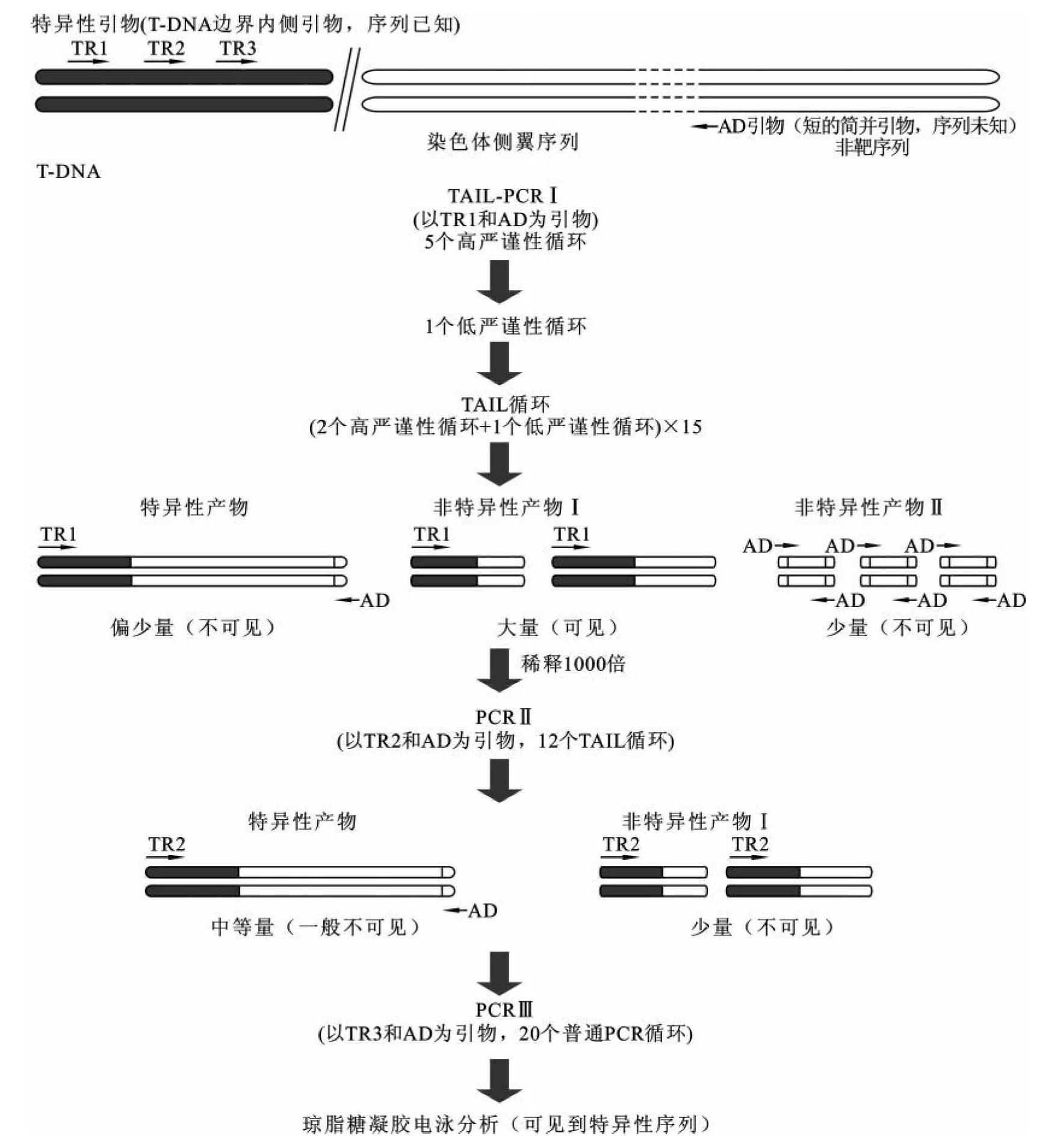
图9-21 TAIL-PCR流程示意图
3.反向PCR法
反向PCR法(inverse or inverted PCR,IPCR)是Triglia等于1988年设计的一种方法,其基本原理(图9-22)是根据已知序列和宿主序列选择合适的酶切位点,对转化体基因组DNA进行完全酶切,酶切产物环化,用一对与已知序列两侧互补的引物进行PCR扩增。对于已知序列来说,引物复性后两引物3'端相背,引物的延伸沿环状分子的未知序列区进行,这与传统的PCR扩增方向相反,故称为反向PCR。该方法在应用时也会存在一些缺陷,如特异性不高、PCR受酶切片段环化效率限制等,因此近些年很少有这方面应用的报道。
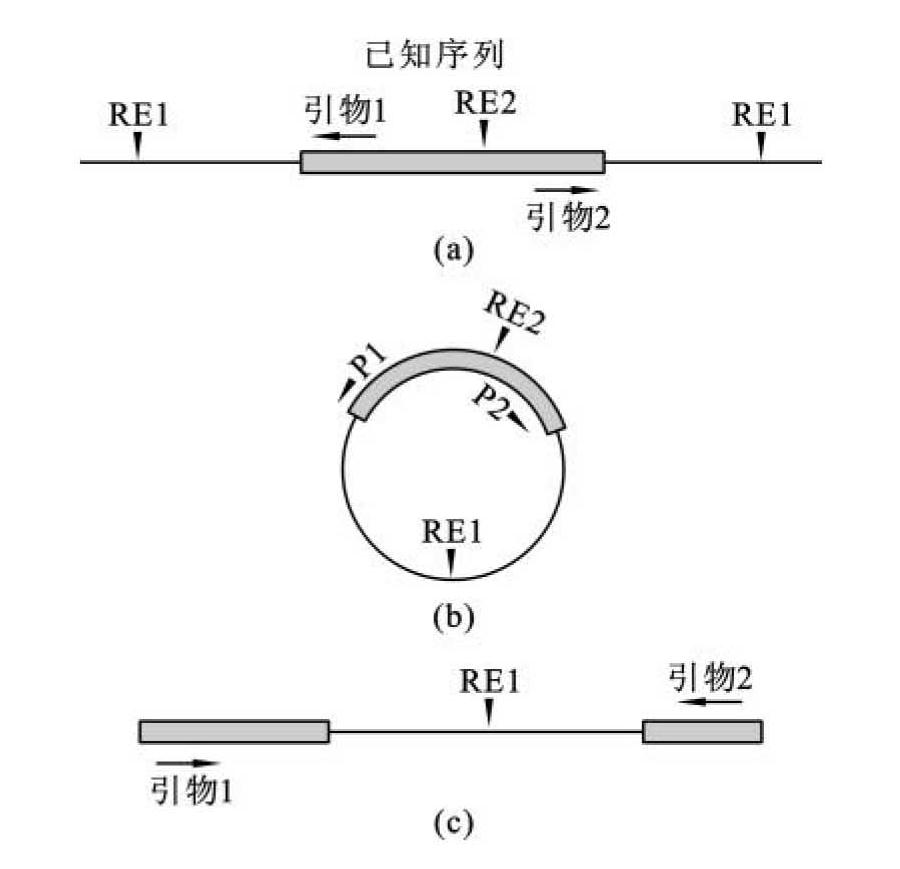
图9-22 反向PCR原理
4.高通量测序法
用高通量测序法可以同时对上百万的核酸分子进行测序,能够全面展示植物的基因组和转录组。对于有参考基因组的植物,只需将转基因植物的全基因组重测序所得到的序列与参考基因组序列比对,通过寻找目的基因,分离出其两端的基因组序列,进而确定目的基因插入位置。对于没有参考基因组的植物突变体,需要先对该物种进行从头测序,组装出参考基因组,而后再对转基因植物进行全基因组重测序,分析外源基因在植物体内的整合情况。