5.5.5 图位克隆
图位克隆(map-based cloning)又称定位克隆(positional cloning),由剑桥大学的Alan Coulson在1986年提出,是在不清楚基因产物结构和功能的情况下,根据基因在染色体上都有稳定的基因座实现的。大片段克隆载体的发展和高密度分子连锁图谱的构建使图位克隆技术的实际应用成为可能,而一些模式生物基因组测序的完成则使在这些生物及同科属生物间图位克隆一个目的基因的时间大为缩短。随着各种分子标记技术和高质量基因组文库构建技术的快速发展,图位克隆已经成为分离生物体基因的一种常规技术。
1.图位克隆的定义和策略
图位克隆是根据目的基因在染色体上的位置进行基因克隆的一种方法。自1992年图位克隆技术首次在拟南芥中克隆到ABI3基因和FAD3基因以来,图位克隆技术在其他快速发展的相关技术支持下迅速发展起来。它是依据功能基因在生物基因组中都有相对稳定的基因座,在利用分子标记技术对目的基因进行精细定位的基础上,用与目的基因紧密连锁的分子标记筛选已构建的DNA文库(如Cosmid、YAC、BAC等文库),构建出目的基因区域的遗传图谱和物理图谱,再利用此物理图谱通过染色体步行、登陆和跳跃的方式获得含有目的基因的克隆,最后通过遗传转化和功能互补实验来验证所获得的目的基因(图5-18)。
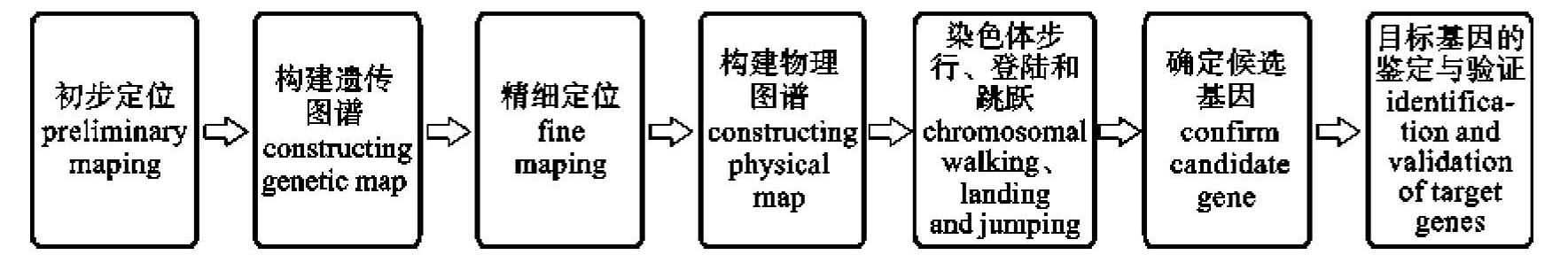
图5-18 图位克隆的策略
2.图位克隆的一般步骤
1)筛选与目的基因连锁的分子标记
利用分子标记技术在一个目标性状的分离群体中把目的基因定位于一定的染色体区域内,对目的基因在染色体上进行初步定位。初步定位目的基因常用近等基因系法(near isogenic lines,NILs)和群组分离分析法(bulk segregant analysis,BSA)。近等基因系是一系列回交过程的产物,理论上近等基因系间除了目的基因及邻近区不同外,其他区段应完全一致。因此,如果在近等基因系中检测出多态性,差异就必定在目的基因及其邻近区域中。但近等基因系法由于基因连锁,在回交导入目标性状基因的同时,与目的基因连锁的染色体片段也随之进入子代中,出现连锁累赘现象,且构建NILs周期过长,从而限制了该法的广泛应用。群组分离分析法由Michelmore等在1991年提出,其原理是将分离群体(F2、BC1、DH系等)中的个体依据研究的目标性状(如抗病、感病)分成两组,在每一组群体中将各个体DNA等量混合,形成两个DNA池(如抗病池和感病池)。由于分组时仅对目标性状进行选择,因此两个池间理论上主要在目的基因区段存在差异。通过初步定位,一般可以筛选出距目的基因的遗传距离一般为5~10 cM的两个侧面连锁的分子标记,这样就可以进行下一步的精细定位工作。
2)目的基因部位的精细定位和作图
精细定位的最终目标是将包含突变基因的遗传间隔缩小到0.5 cM甚至更小。显然用于作图的定位群体越大,就越能精确地定位突变基因。一般需要包含3000~4000个生物个体的定位群体来精确地定位目的基因。如果目的基因是在着丝粒附近,1 cM相当于1000 kb左右,再加上着丝粒附近染色体重组率低,要想获得与目的基因紧密连锁的分子标记,就必须建立更大的定位群体,这样可以为后面的染色体步行节省时力。精细定位工作是图位克隆一个基因过程中最为耗时耗力的步骤,也是限速的一步。增加一个已知区域内的分子标记的方法如下:首先整合已有的遗传图谱,将各种遗传图谱中的分子标记整合到一块,可以提高分子标记的密度;其次可以增加新的分子标记,如在水稻、拟南芥这些已经测序的生物上,可以利用目的基因两侧的DNA序列和分子标记设计软件(SSRHunter、Primer Premier 5.0等)来大量设计新的分子标记;此外,也可以利用比较基因组的共线性从其他同科属的生物上获得分子标记。用这些分子标记对建图群体进行精细定位,以期找到与目的基因更紧密连锁的分子标记甚至共分离的分子标记。对定位群体较大,需要对单株进行分析,提取大量DNA的问题,可根据Churchill等1993年提出的DNA混合样品作图的方法进行实验设计。DNA混合样品作图是指把大群体中所需进行分子标记多态鉴定的单株(个)分成若干组(可5~20个单株一组),以组为单位提取DNA,形成一个组内混合的DNA池,用精细定位的分子标记对混合的DNA池进行分析,根据所有池中的分子标记与目的基因发生的重组数来确定目的基因附近分子标记的顺序。混合样品作图大大地提高分子标记分析效率,减少了DNA提取的工作量,有利于扩大群体,加速克隆进程。
通过具有多态性的分子标记对作图群体的分析,根据染色体上分子标记与目的基因间的重组率构建出目的基因附近的遗传图谱。但分子标记与目的基因之间的距离是按照实际的碱基数来计算的,所以物理图谱才是真正意义上的基因图谱,它会因不同染色体区域基因重组值不同而造成与遗传距离的差别。物理图谱的种类很多,有染色体分带图、限制酶切图谱、跨叠克隆群、DNA序列图谱等。限制酶切图谱是用几种限制性核酸内切酶消化DNA,通过电泳检查限制性片段长度的办法确定它们的排列顺序;对于较大的基因组,可以利用稀有切点限制性核酸内切酶和脉冲电脉。跨叠克隆群的制作则需具有一定容量的大片段基因组文库。比较各个克隆的插入片段,将它们排列成与原来在染色体中的顺序一样的连续克隆群,即跨叠克隆群。荧光原位杂交技术(Fiber-FISH)使FISH技术的分辨率接近其理论值1 kb(相当于约0.14 μm的DNA纤丝),即光学显微镜的识别范围内。Fiber-FISH适宜用基因组的数量作图,而且由于可以在荧光显微镜下同时观察几种探针的位置和顺序,将有效地排除染色体步行过程中经常遇到的重复序列带来的困难。所有这些技术的发展和出现都为图位克隆基因奠定了坚实的基础。(https://www.daowen.com)
3)染色体步行、登陆和跳跃
染色体步行(chromosome walking)是通过逐一克隆来自染色体基因组DNA的彼此重复的序列,而慢慢地靠近目的基因,开始步行的克隆可以是已知的基因、RFLP、RAPD或其他已鉴定的分子标记,用它来杂交筛选大片段DNA文库中的阳性克隆,找出与目的基因两侧连锁最紧密的分子标记所在的大片段克隆,接着分别以两侧分子标记所在的克隆为起点进行染色体步行,逐步靠近目的基因。以大片段克隆的末端为探针,筛选基因组文库,鉴定和分离出邻近的基因组片段的克隆,再将这个克隆的远末端作为探针重新筛选基因组文库;继续这一过程,直到获得具有目的基因两侧分子标记的大片段克隆或跨叠克隆群。当遗传连锁图谱指出基因所在的特定区域时,即可取回需要的克隆,获得目的基因。
染色体步行在实际运用中的主要困难是当在克隆的一端遇到大量重复的DNA序列时,步行的方向会被打乱;另外,当步行必须经过一个间隙时,步行的过程就会被打断。这些都会造成图位克隆的失败。为了克服这些困难,人们相继提出了染色体登陆、跳跃和连接等方法。染色体登陆是找出与目的基因的物理距离小于基因组文库插入片段的平均距离的分子标记。一般找到与目的基因共分离的分子标记,通过这样的分子标记筛选文库可直接获得含有目的基因的克隆,完全避开染色体步行的过程。染色体跳跃、连接分别是使用一个识别位点很少的酶和一个识别位点很多的酶构建跳跃文库、连接文库。跳跃文库的插入片段是大片段克隆末端经过双酶切的部分,由同样的文库进行克隆。连接文库的插入片段是由切点较少的酶产生的,具有切点较少的那个酶的识别位点。在染色体步行的过程中,交替应用两个文库进行跳跃和连接,最终逼近目的基因。
4)目的基因的鉴定与验证
得到的目的基因所在的小片段克隆有可能含有多个开放阅读框,从这些开放阅读框中鉴定目的基因是图位克隆技术的最后一个关键环节。常用的方法是用含有目的基因的大片段克隆(如BAC克隆或YAC克隆)去筛选cDNA文库,并查询生物数据信息库,待找出候选基因后,把这些候选基因进行下列分析以确定目的基因:①用精细定位法检查cDNA是否与目的基因共分离;②检查cDNA时空表达特点是否与表型一致;③测定cDNA序列,查询数据库,以了解该基因的功能;④筛选突变体文库,找出DNA序列上的变化及其与功能的关系;⑤进行功能互补实验,通过转化突变体观察突变体表型是否恢复正常或发生预期的表型变化。功能互补实验是最直接鉴定基因的方法。利用RNA干扰(RNAi)也可有效地确定目的基因。
3.图位克隆技术的局限性
利用图位克隆法克隆基因不仅需要构建完整的基因组文库,建立饱和的分子标记连锁图和完善的遗传转化系统,而且还要进行大量的测序工作,所以对基因组大、标记数目不多、重复序列较多的生物采用此法不仅投资大,而且效率低。因而图位克隆法仅应用在人类、拟南芥、水稻、番茄等生物上。此外,在分析发生的变异时,最有可能遇到的复杂情况是一个给定的性状由不止一个的基因位点控制。例如,在拟南芥Kashmir-1(有抗性的)和Columbia(敏感的)株系之间的杂交实验中,粉状霉菌抗性基因至少涉及三个遗传位点,它们是以附加的方式起作用的。对这些抗性基因中的任何一个作精细定位都要求降低作图群体的遗传复杂性,如创造只有一个位点保持多态性的重组近交系。因此,当影响这些性状的自然或者诱导的突变被定位时,如第二位点修饰成分干扰这些分析,将使图位克隆此类基因变得非常困难。染色体上位点的物理距离和遗传距离的比值是变化的,通常这种变化是比较小的,对作图的分辨率也只有较小的影响。但如果要定位的基因位于重组被严格限制的着丝粒附近,精细定位的努力就有可能无效。对常染色体序列1%重组的遗传距离相当于100~400 kb的物理距离,然而着丝粒区域1%重组的遗传距离相当于1000~2500 kb的物理距离,在现存的物理图谱中很少有着丝粒区域被覆盖。这些都使图位克隆的染色体步行变得不可能。
4.图位克隆技术的展望
进入21世纪,生命科学取得了前所未有的发展,一系列模式生物的全基因组序列被测出,与生物研究相关的理论、方法日新月异。图位克隆是较为通用的基因获得技术,在理论上适用于一切基因。基因组研究产生了很多便宜但功能强大的工具,同时也有大量的信息被收集在免费的数据库中(高精度遗传图谱、大尺度物理图谱、大片段基因组文库,甚至基因组全序列),这为图位克隆的广泛应用提供了条件。现在,图位克隆已经成为分离基因的常规方法。有越来越多的生物通过图位克隆技术克隆到基因,如拟南芥、水稻、番茄等。图位克隆基因不仅适用于单基因克隆,对于那些由多基因控制的数量性状的定位同样适用。农作物中许多重要的农艺性状都是数量性状,受多个基因(如花时、籽粒大小、生理节律、次生代谢等)的控制。目前,利用图位克隆分离此类基因,主要是通过不断的回交构建QTL(quantitative trait locus,数量性状座位)近等基因系以减少分离群体的遗传背景,来实现对单个QTL遗传效应的分析,并对QTL进行精细定位。随着越来越多的QTL被定位到分子标记连锁图上,利用图位克隆技术克隆单个贡献率较大的基因来改良作物已成为可能。